Organic molecules with extended π-conjugation frameworks are emerging as promising candidates for active media in nanoscale optoelectronic applications. Benzodichalco-genophene (BDC) derivatives, in particular, exhibit rigid planar geometries and tunable electronic properties, making them attractive for use in single-molecule laser devices. This study theoretically examines the structural, electronic, optical, and charge transport properties of several BDC molecules using advanced computational methods. Geometry optimizations were conducted with the Perdew Burke Ernzerhof (PBE) functional via the SIESTA package, while electronic properties were evaluated at the B3LYP/3-21G level. Time-dependent density functional theory (TD-DFT) was employed to simulate optical absorption spectra, and the GOLLUM code was used to model charge transport through molecular junctions based on non-equilibrium Green’s function formalism. The findings reveal that increasing molecular length narrows the highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) gap, enhances orbital delocalization, and improves electron transmission. Optical simulations revealed red-shifted absorption peaks and increased oscillator strengths, indicating enhanced light-matter interactions. Furthermore, density of states analysis confirmed the transition from HOMO- to LUMO-dominated transport with greater conjugation. Overall, BDC derivatives show strong potential for integration into molecular-scale lasers and optoelectronic devices, paving the way for future experimental and technological advancements.
Introduction
Carbon-based molecular junctions, such as carbon nanotubes and graphene nanoribbons, have become foundational elements in the evolution of nanoscale electronics due to their outstanding electrical conductivity, mechanical robustness, and thermal stability [1–3]. One of the pivotal goals in this domain is to identify and optimize molecular-scale semiconductors that can bridge nanogaps between metal electrodes while maintaining desirable charge transport properties [4].
Among the various candidates explored for this purpose, benzodichalcogenophenes (BDCs) have attracted significant attention due to their unique structural and electronic characteristics. These compounds are composed of five-membered heterocycles fused with six-membered aromatic rings and contain heteroatoms such as sulphur (S) and oxygen (O), which enable tunability in electronic properties. Their rigid, planar π-conjugated frameworks facilitate delocalized electron transport, minimize structural deformation, and offer favourable highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) alignments for electronic applications [5, 6].
The variation in molecular structure, specifically changes in conjugation length and heteroatom substitution, directly affects key optoelectronic properties, including the HOMO–LUMO energy gap, charge carrier mobility, and oscillator strength. An increase in π-conjugation generally leads to a reduction in the HOMO–LUMO gap, enhancing the material's ability to interact with light and improving its potential performance in charge transport and photonic applications [7, 8]. These structure–property relationships are most effectively probed using computational techniques such as density functional theory (DFT) and time-dependent DFT (TD-DFT), which offer a predictive insight into the impact of geometric modifications on electronic behaviour [9, 10].
The potential of organic π-conjugated molecules in photonic applications, particularly as active gain media in single-molecule lasers, has gained increasing momentum. Compared to inorganic laser materials, organic systems offer distinct advantages, including solution processability, structural diversity, and tunable emission spectra [11]. When excited optically or electrically, these molecules can undergo radiative transitions necessary for lasing, thereby enabling the development of low-cost, tunable, and miniaturized photonic components [12, 13].
Several classes of organic molecules have demonstrated promising lasing capabilities, including oligothiophenes, diketopyrrolopyrroles (DPPs), polyfluorenes, and BODIPY-based dyes [14–17]. These systems typically exhibit high photoluminescence efficiency, strong oscillator strengths, and tunable optical properties. However, many are hindered by issues such as aggregation-induced quenching (AIQ), poor environmental stability, or structural non-planarity under excitation [18, 19]. In contrast, BDC derivatives offer structural rigidity and electronic tunability, positioning them as promising, though relatively underexplored, candidates for next-generation laser-active media [20].
In response to the growing demand for predictive modelling in optoelectronic material design, numerous studies have utilized DFT to investigate organic and hybrid compounds. For example, Maadhu and Gandhiraj [21] applied DFT to bis-morpholinium zinc bromide crystals, uncovering notable optical band gaps and nonlinear optical behaviour. Maadhu et al. [22] explored a mercury-based metal-organic framework (BMMC), correlating DFT-derived frontier orbitals with experimentally observed absorption spectra. Maadhu et al. [23] analysed pyridinium-based organic crystals, revealing efficient photon absorption and notable stability. Maadhu et al. [24] also examined morpholinium bromide crystals and identified key electronic transitions relevant for optical applications using DFT.
While these studies underscore the effectiveness of DFT in elucidating optical and electronic behaviours across diverse materials, most focus on non-conjugated systems or metal-organic hybrids. The present investigation addresses this gap by targeting π-conjugated BDC derivatives to elucidate how systematic variation in molecular length influences optoelectronic responses.
This work adopts a combination of DFT, TD-DFT, and non-equilibrium Green’s function (NEGF) formalism to assess the structural, optical, and transport properties of BDC molecules. The methodology includes geometry optimization, electronic structure analysis, simulation of absorption spectra and oscillator strengths, and modelling of electron transmission through molecular junctions using the GOLLUM code [25]. Theoretical insights derived from these calculations aim to inform the design of BDC-based systems for advanced optoelectronic and photonic applications.
Materials and computational methods
To examine the optoelectronic behaviour of single-molecule junctions based on BDC, the authors implemented a sequence of computational methods that integrate the NEGF formalism with DFT. These techniques are indispensable for assessing molecular geometries, electronic structures, charge transport mechanisms, and optical absorption behaviour at the nanoscale [26,27].
A combination of first-principles quantum mechanical methods was employed to investigate the optoelectronic performance of BDC-based single-molecule junctions [28].
Molecular structure optimization
Initial geometries of the BDC molecules (designated as 1B through 5B) were constructed and fully optimized in the gas phase using the SIESTA simulation package [29].
Structural relaxation was performed using the generalized gradient approximation (GGA) with the Perdew Burke Ernzerhof (PBE) exchange-correlation functional [30]. A
double-zeta polarized (DZP) basis set was applied to describe atomic orbitals and a real-space grid with a cut-off
energy of 250 Ry was used [31]. The geometry
optimization was considered complete when the maximum force on any atom fell below 0.01 eV/A.
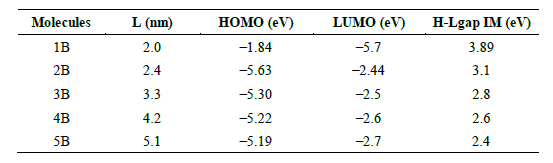
All molecules maintained their rigid, planar π‑conjugated backbones after relaxation, an essential feature for electronic stability in molecular junctions [32]. Molecular lengths were extracted for each relaxed geometry and are presented in Table 1.
The B3LYP hybrid functional combined with a 6-31G basis set was used to perform electronic structure analysis. This degree of theory reliably describes the border molecular orbitals, specifically HOMO and LUMO [33]. The HOMO–LUMO energy gaps were calculated and orbital density plots were generated to visualize electron delocalization across the molecular backbone, which plays a crucial role in charge transport efficiency [34].
Transport simulations
To study the transport behaviour of the BDC molecules, each optimized structure was positioned between two 35‑atom gold electrodes arranged in a pyramidal configu-ration. The resulting molecular junctions were analysed using the GOLLUM quantum transport code [35]. Transport properties such as transmission spectra, electron tunnelling probabilities, and zero-bias conductance were calculated using the NEGF approach. This method accurately models charge flow through a molecule under open boundary conditions, replicating the behaviour of real-world electronic devices [36].
Optical property analysis
TD-DFT calculations at the B3LYP/6-31G level were used to determine the UV-visible absorption spectra of all BDC molecules. These calculations yielded excitation energies, maximum absorption wavelengths (λmax), and oscillator strengths [37]. The main transitions were identified as HOMO–LUMO in character, with longer molecules showing stronger absorption and larger oscillator strengths indicative of the enhanced light-matter interaction [38]. This trend supports their suitability as active laser media in molecular-scale optoelectronic systems [39].
Results and discussion
This section presents a comprehensive analysis of the structural, electronic transport and optical properties of the BDC-based molecular systems (1B to 5B). The authors’ findings illustrate how molecular length and conjugation influence electron transport and light-matter interactions, confirming the potential of these systems for nanoscale optoelectronic and photonic applications [40].
Structural and geometrical properties
The relaxed molecular geometries, depicted in Fig. 1.
reveal a progressive extension of the BDC molecular backbone across the series (1B to 5B), while consistently preserving a planar π-conjugated structure. This structural planarity is essential for maintaining π-orbital overlap, enabling continuous delocalization of charge carriers along the molecular axis, which is a prerequisite for efficient electron transport in molecular junctions [41,42]. The rigidity conferred by the fused aromatic and heterocyclic rings minimizes conformational distortions, even as the conjugated length increases. This characteristics is particularly beneficial under electrical bias, as fluctuations in molecular geometry are known to disrupt conductance pathways and induce scattering losses [43,44].
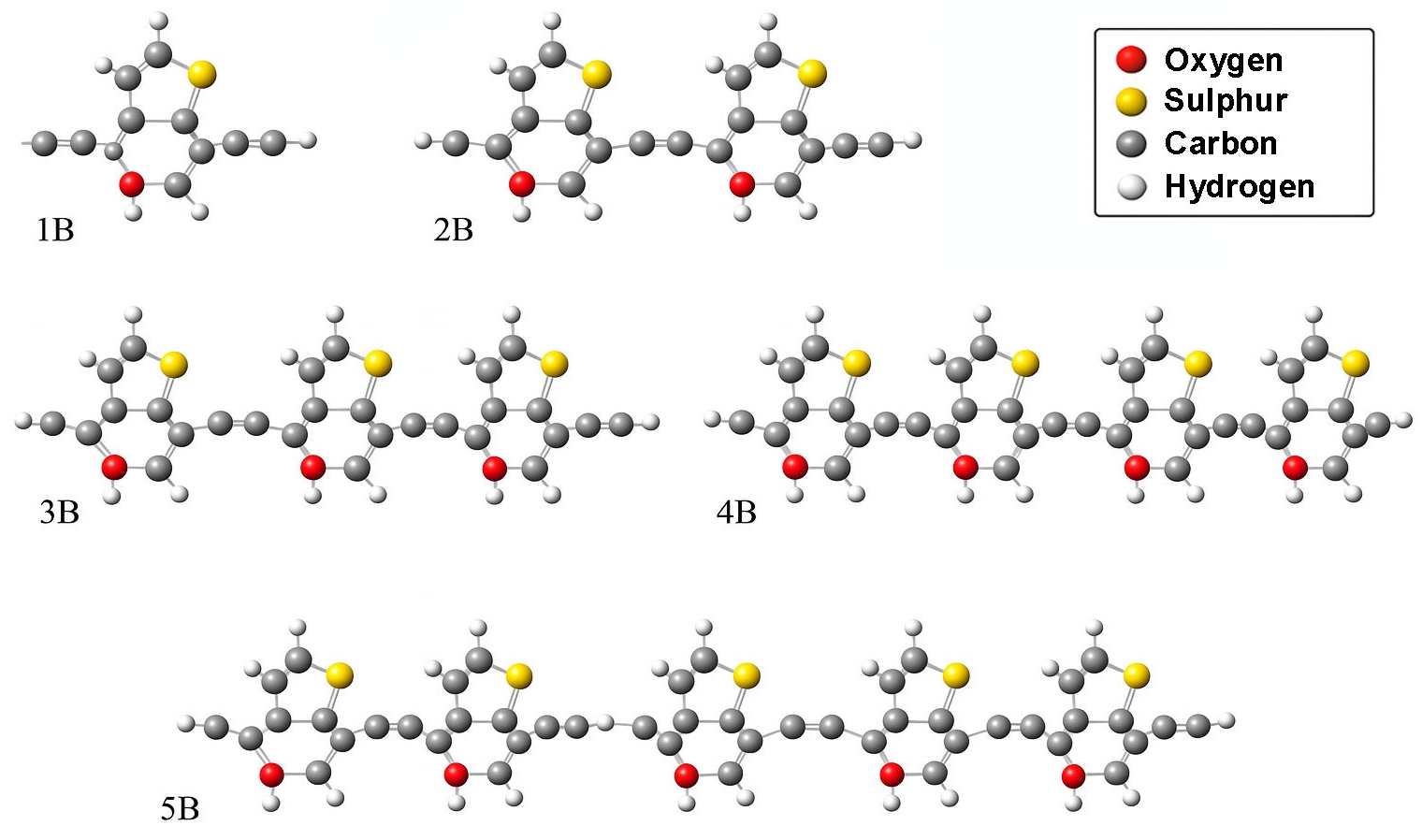
Fig. 1.Optimized gas-phase geometries of five BDC derivatives, labeled 1B through 5B. Each structure consists of alternating sulphur
(yellow) and oxygen (red) heteroatoms fused into π-conjugated aromatic systems. The molecular length increases systematically
from 2.0 nm (1B) to 5.1 nm (5B) with the addition of BDC repeat units
The optimized lengths of the molecules range from 2.0 nm (1B) to 5.1 nm (5B), as shown in Table 1. Despite this increase in molecular length, no significant deviation from planarity is observed, indicating high structural resilience and mechanical stability. Such preservation of moleculor integrity across different chain lengths ensures uniform coupling to metal electrodes, reduces junction instability, and supports reproducible charge transport performance in device architectures [45]. This observation
confirms that the BDC framework is inherently well-suited for electronic applications that require structural robustness and coherent charge propagation at the molecular scale.
Electronic structure properties
To gain a deeper understanding of the electronic behaviour of BDC molecules and their role in charge transport within manalysis of the frontier molecular orbitals using DFT at the B3LYP/3-21G level of theory, which provides a reliable balance between computational efficiency and accuracy for conjugated organic systems [43,46].
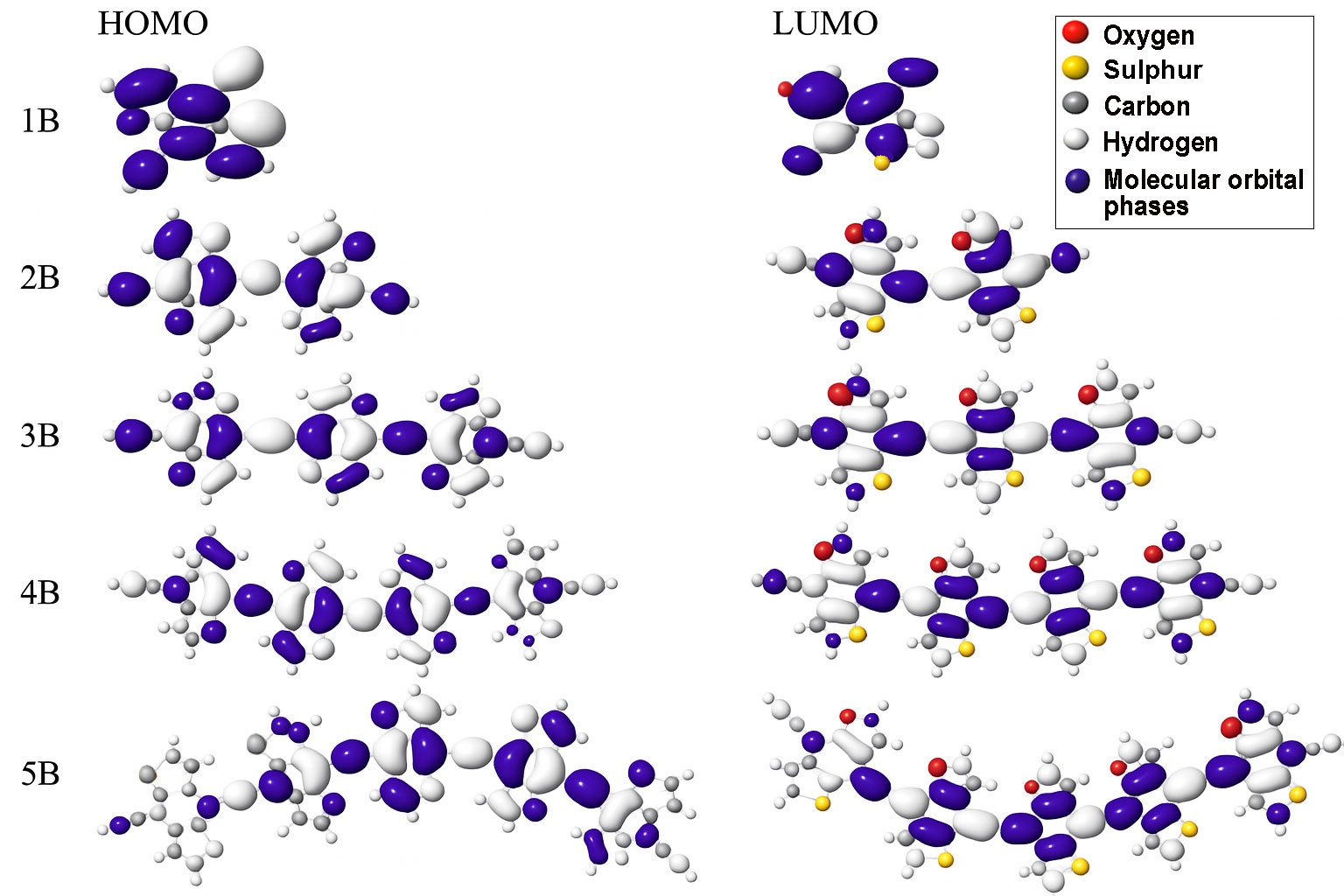
The results revealed that the HOMOs are primarily localized on the heteroatoms, particularly S and O, as well as portions of the aromatic backbone. In contrast, the LUMOs appear more delocalized along the conjugated carbon framework, especially across C–C bonds [47]. This contrast in orbital distribution suggests that charge transport in these molecules is likely dominated by the HOMO, especially in shorter chains, where the HOMO is more effectively aligned with the electrode interfaces [48].
The spatial distribution of these frontier orbitals is visualized in , highlighting the HOMO localization and LUMO delocalization across the molecular frameworks.
In molecules featuring pyridine anchor groups, the LUMO shows a slightly higher charge density than the HOMO. However, its lower orbital weight implies a limited contribution to electron transport under standard conditions [49]. This difference in orbital composition suggests that the likelihood of junction formation may be high, although it may be accompanied by junction instability in specific configurations due to electronic asymmetry [50].
Furthermore, in short-chain BDC molecules, the contrast between HOMO localization primarily on heteroatoms and adjacent aromatic rings and the delocalization of the LUMO across the conjugated carbon backbone plays a key role in defining charge transport behaviour. The localized HOMO enables stronger electronic coupling with the electrode interface, enhancing hole injection and facilitating HOMO-mediated transport. Meanwhile, the more delocalized but energetically misaligned LUMO contributes less to electron transport in short chains due to its reduced spatial overlap and greater energy offset from the Fermi level. As the molecular length increases, this asymmetry diminishes, promoting LUMO-mediated electron conduction. This evolution reflects a length-dependent transition in transport pathways, reinforcing the structural tunability of BDC systems for molecular electronics [4,42].
Fig. 2.Visual representation of the HOMO and LUMO for a series of BDC derivatives labeled 1B through 5B.
The computed HOMO–LUMO energy gaps, presented in Table 2, show a clear and consistent decrease with increasing molecular length from 3.89 eV in the shortest molecule (1B) to 2.4 eV in the longest (5B). This trend can be attributed to the quantum size effect, where longer conjugation paths enhance π-delocalization and reduce excitation energies [48]. This narrowing of the electronic bandgap is favourable for semiconducting behaviour and optoelectronic applications [51].
All molecules exhibited HOMO localization and LUMO delocalization, supporting efficient intramolecular charge separation [52]. Furthermore, the distribution of aromatic rings and molecular symmetry appear to influence the energy and alignment of the frontier orbitals. These findings confirm that BDC-based molecules possess tunable electronic properties, making them promising candidates for application in molecular-scale electronic and optoelectronic devices [32].
To assess the charge transport behaviour of BDC molecules in a realistic device environment, each optimized structure was positioned between two pyramidal gold electrodes composed of 35 atoms. The resulting molecular junctions were analysed using the NEGF formalism as implemented in the GOLLUM quantum transport code [35]. This method
provides a rigorous approach to simulate electron transmission through molecular systems under open
boundary conditions, closely mimicking experimental transport setups [36].
The transmission spectra computed at zero bias revealed that the conductance of the molecules generally improves with increasing molecular length, contrary to the typical decrease observed due to increased tunnelling distance [53]. This enhancement is attributed to enhanced conjugation, which promotes delocalization of the frontier orbitals, particularly the LUMO, along the molecular backbone [54]. As conjugation extends, orbital overlap with the electrode states improves, leading to more favourable alignment with the Fermi level [27].
In shorter BDC molecules, transport was predominantly HOMO-mediated, as the HOMO orbitals were energetically closer to the Fermi level of the electrodes and more spatially localized on the anchoring groups [54]. However, as the molecular length increased, a shift in the dominant transport channel toward the LUMO was observed. This shift suggests a lengthdependent transition in the charge transport mechanism from hole-like to electron-like conduction [56]. These findings highlights the curitical role of molecular architure in determining the transport pathways and underscore the potential of BDC derivatives for use in tunable nanoelectronic devices [57].
Optical absorption properties
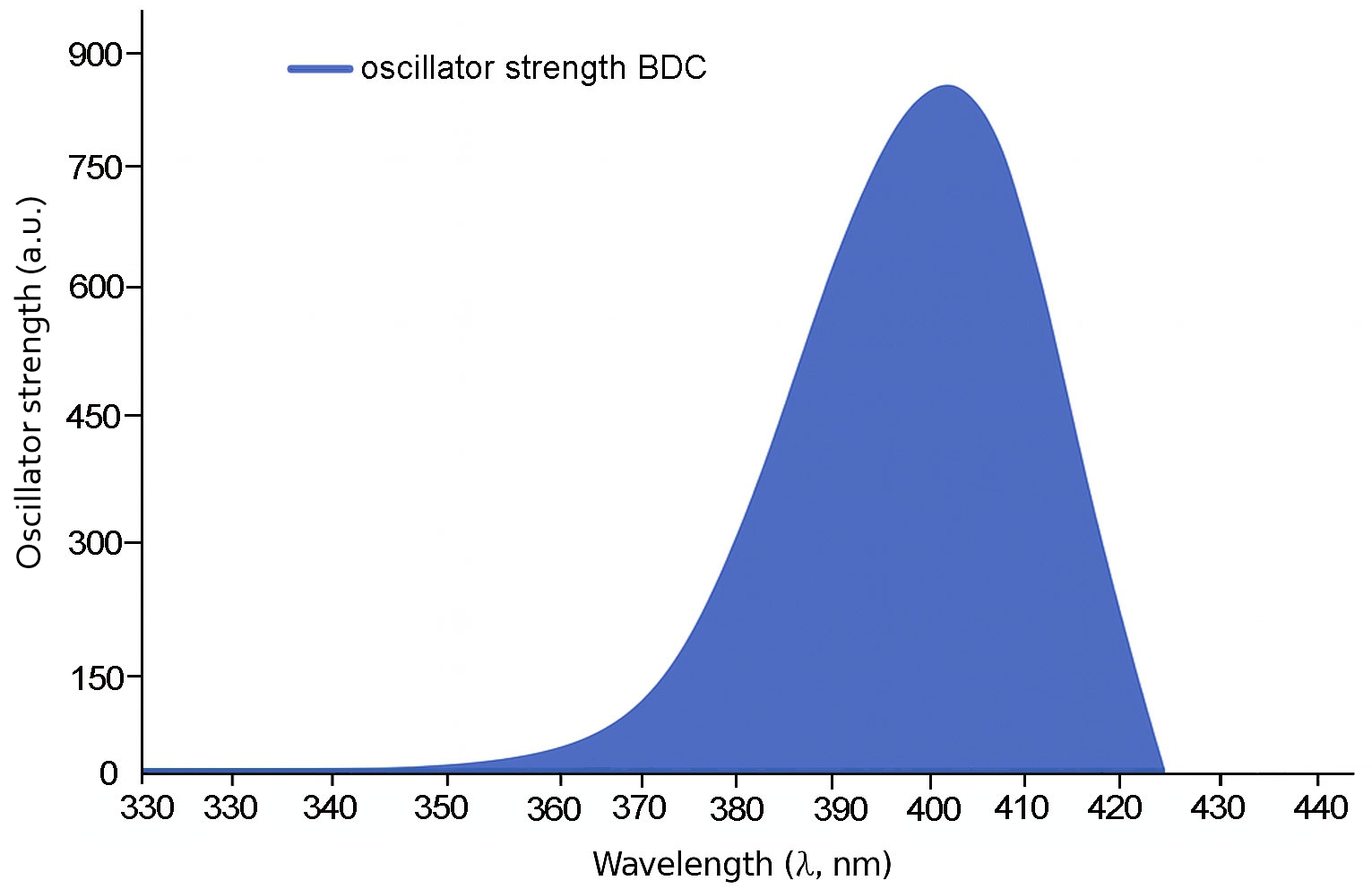
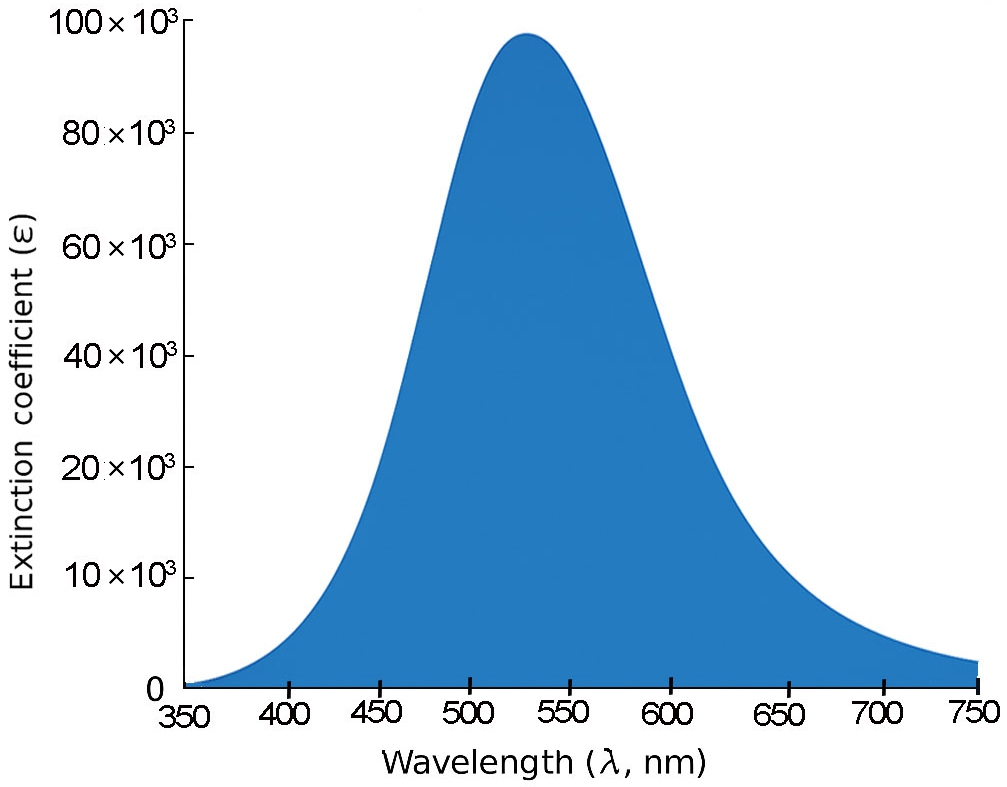
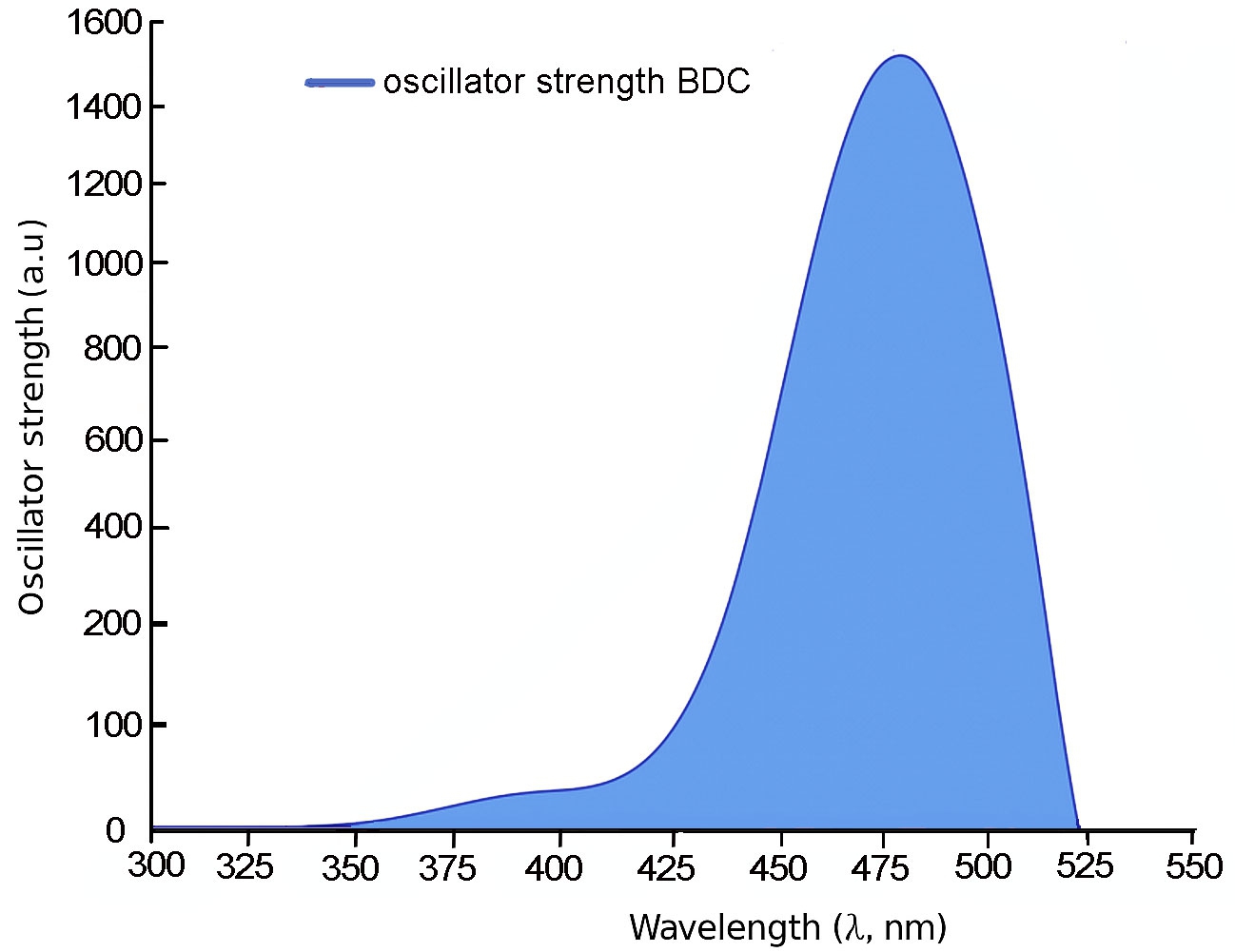
The optical properties of the BDC molecular series were examined using TD-DFT at the B3LYP/3-21G level of theory [55, 58]. The simulated UV-visible absorption spectra and oscillator strength profiles for each molecule (1B to 5B) are presented in Figs. 3 through 7. These plots illustrate the extinction coefficient (ε) and oscillator strength as a function of wavelength and provide insight into the influence of conjugation length on photophysical behaviour [59].
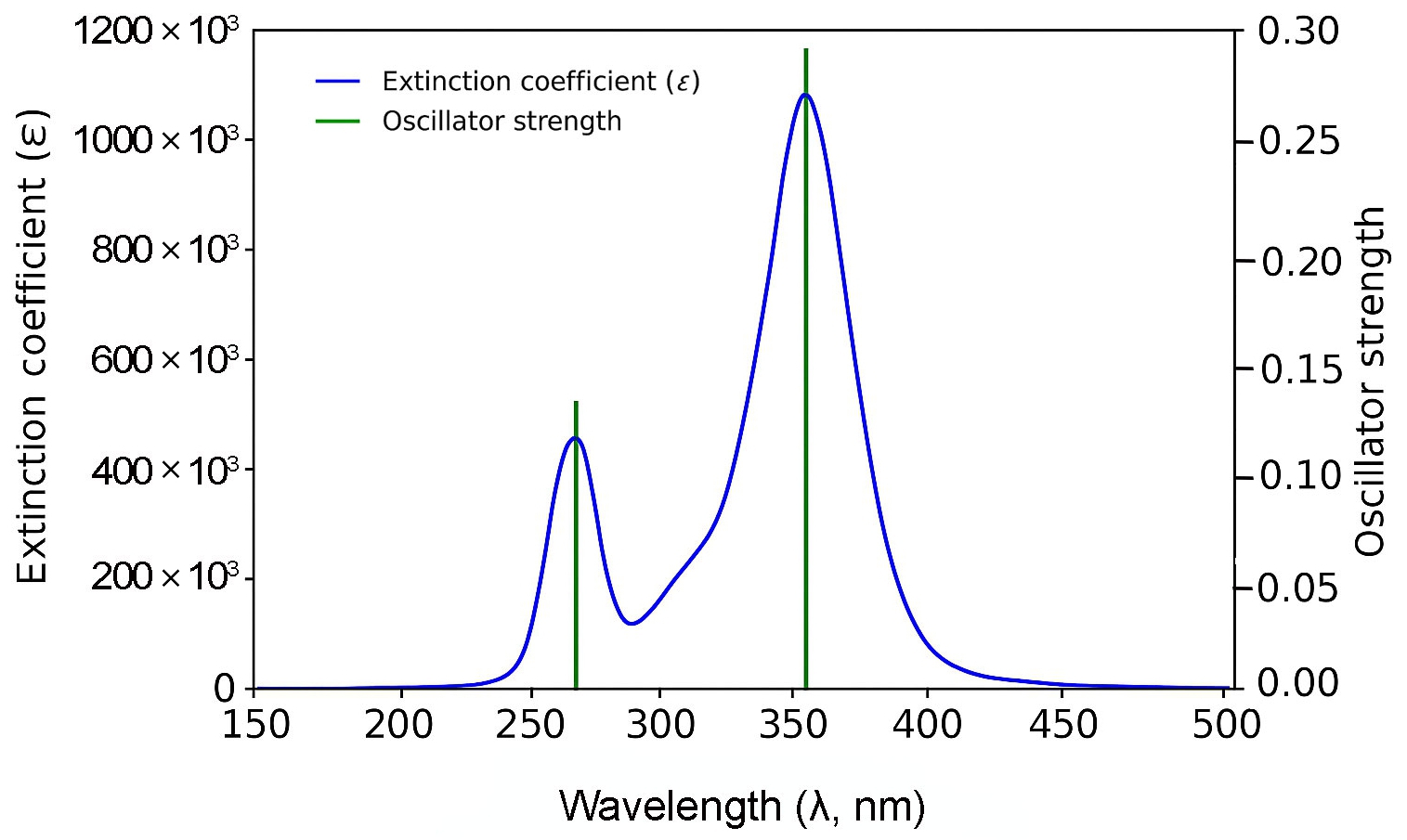
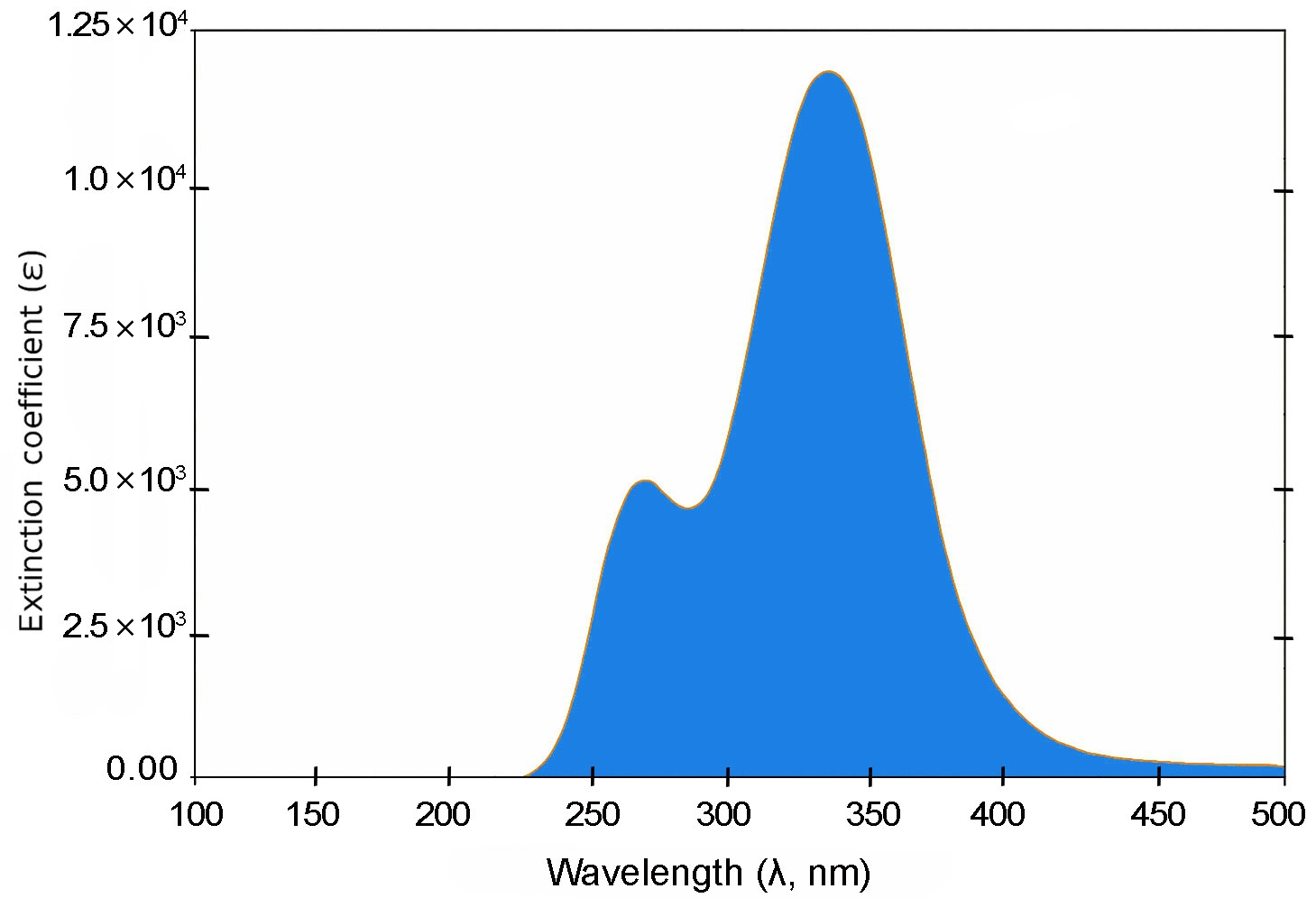
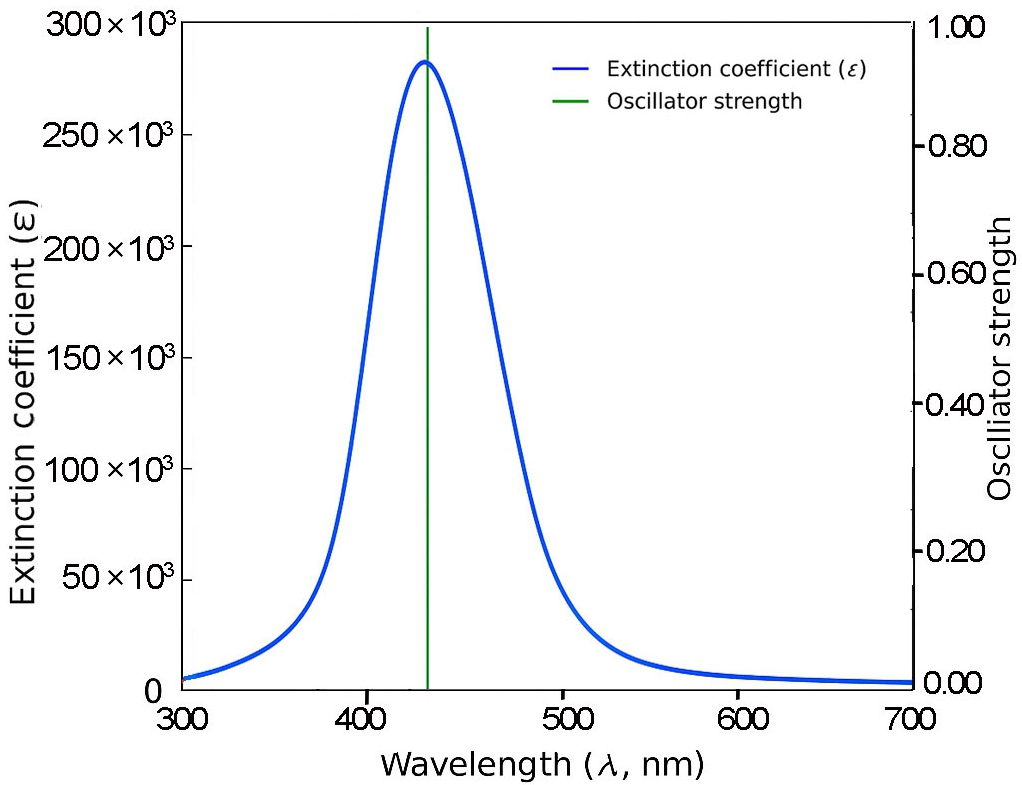
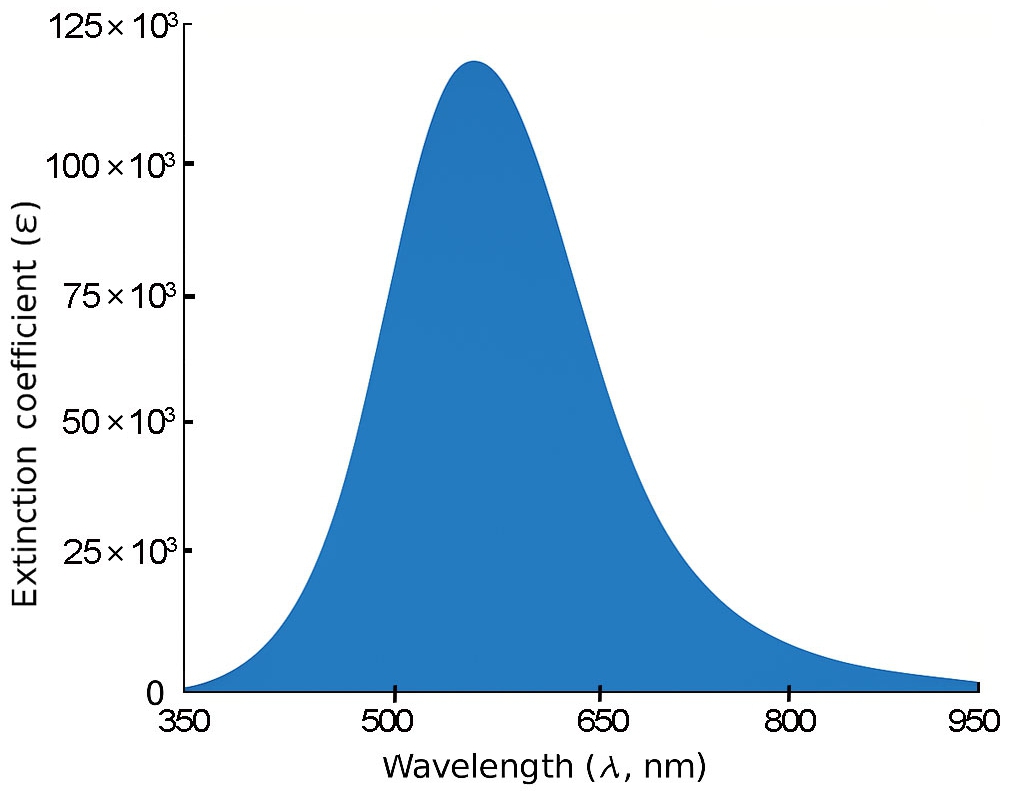
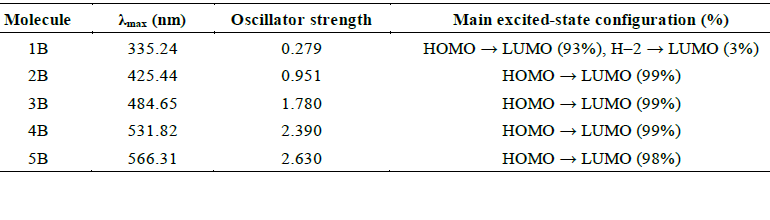
Fig. 3 shows that the shortest molecule, 1B, exhibits a primary absorption peak at approximately 335 nm with a relatively modest oscillator strength of 0.279, indicating limited π-conjugation. In contrast, molecule 2B (Fig. 4) displays a red-shifted absorption peak near 425 nm and a substantially higher oscillator strength of 0.951, reflecting enhanced electronic delocalization.
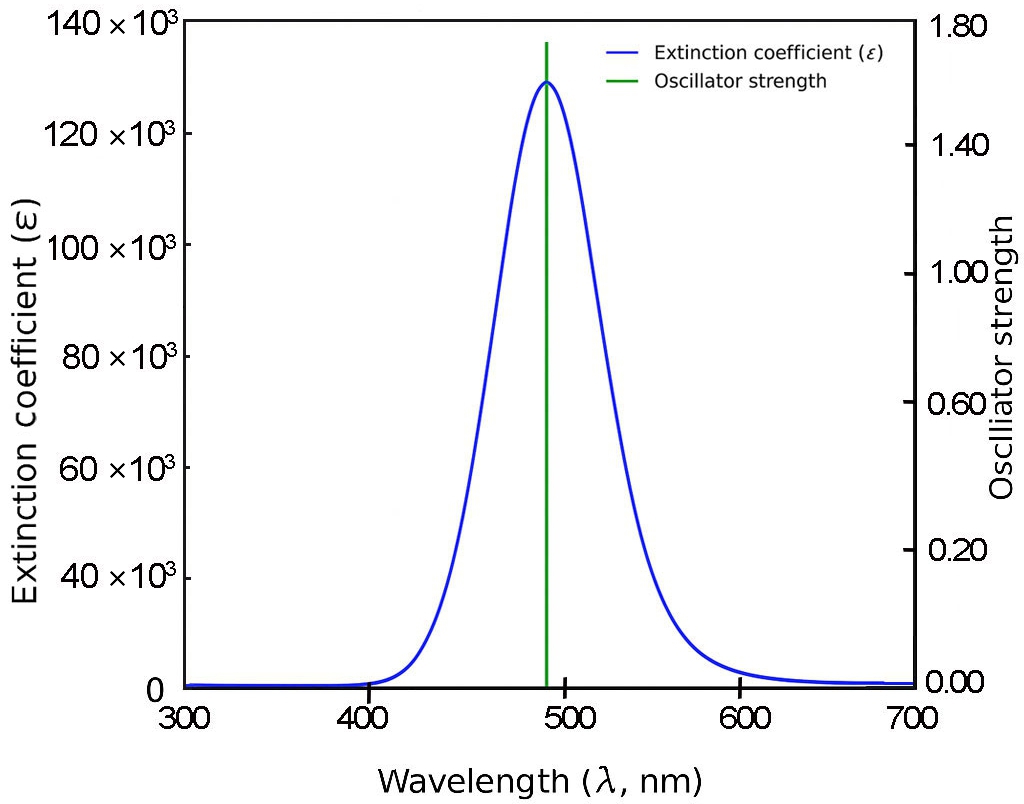
Fig. 3. UV-visible absorption spectrum and oscillator strength profile of BDC molecule 1B. The top panel shows the calculated extinction
coefficient (ε, blue) and oscillator strength (green) as functions of wavelength, while the bottom panels present the smoothed
spectral curves individually.
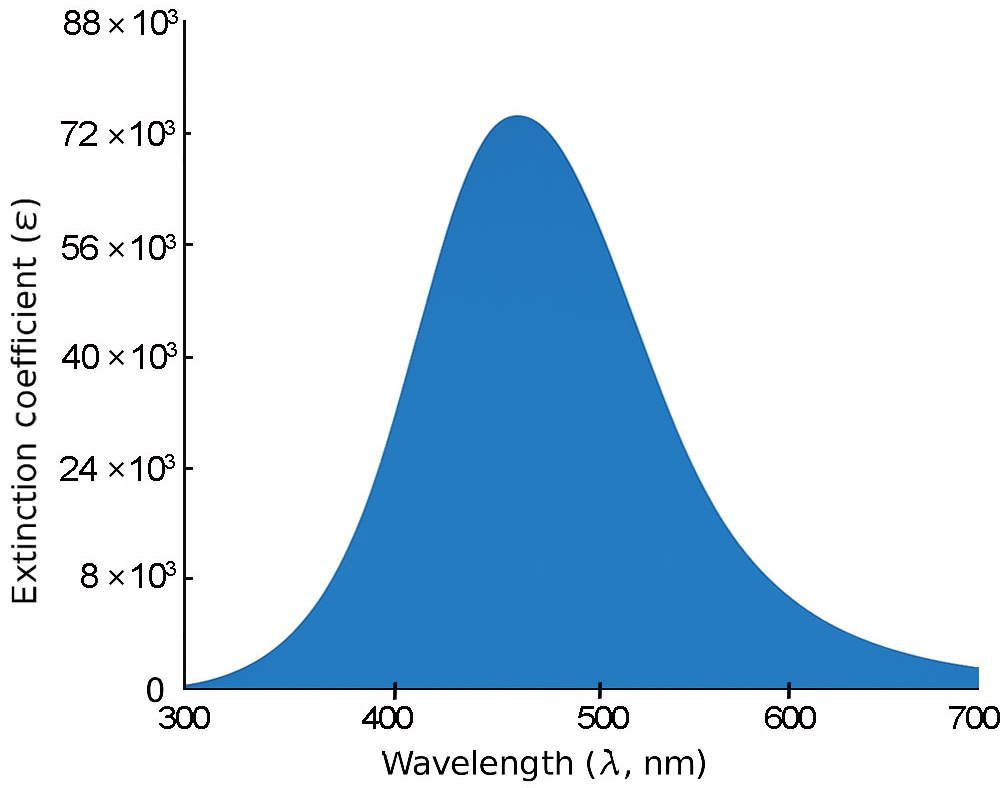
Fig. 4. UV-visible absorption spectrum and oscillator strength profile of BDC molecule 2B. The top panel displays the computed extinction
coefficient (ε) and oscillator strength as functions of wavelength, while the bottom panels illustrate the broadened spectral
responses.
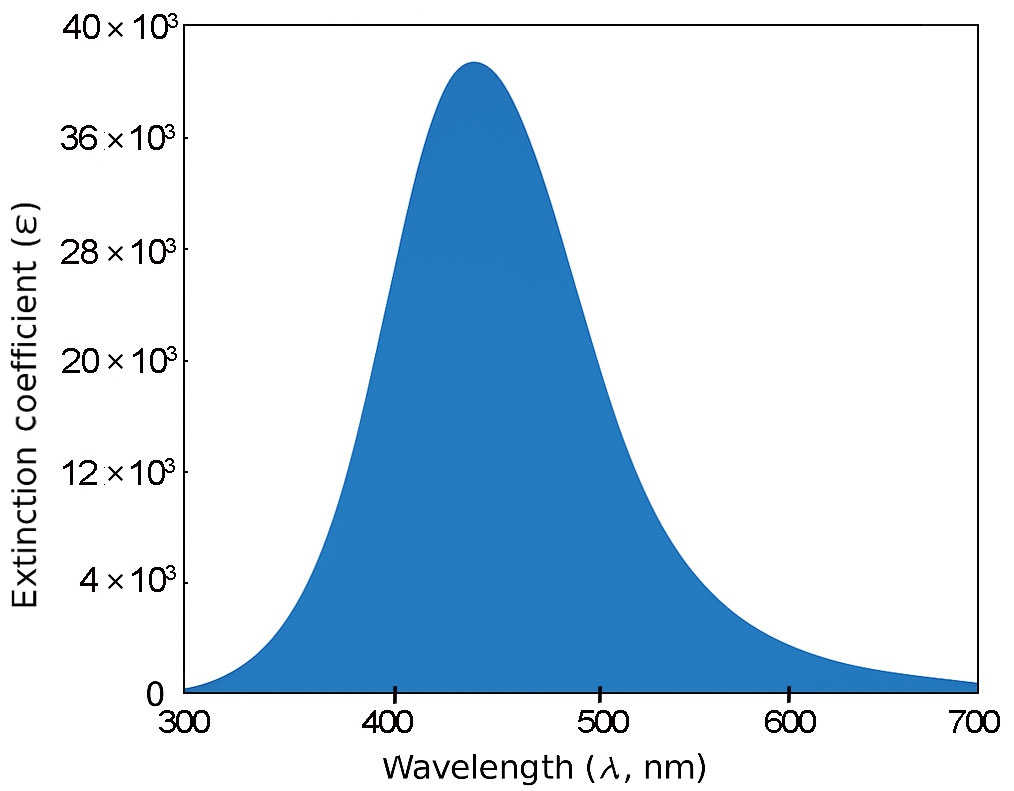
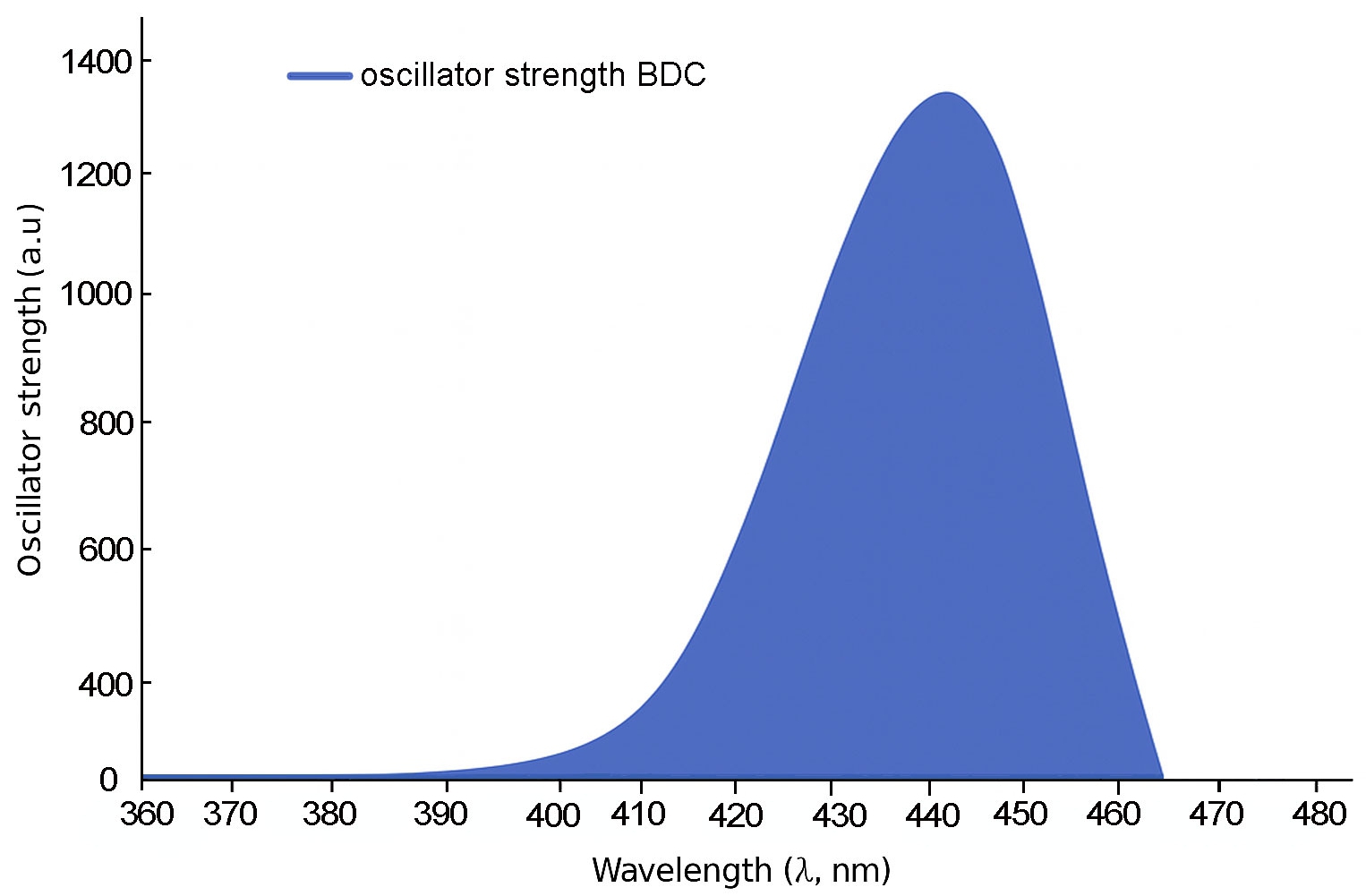
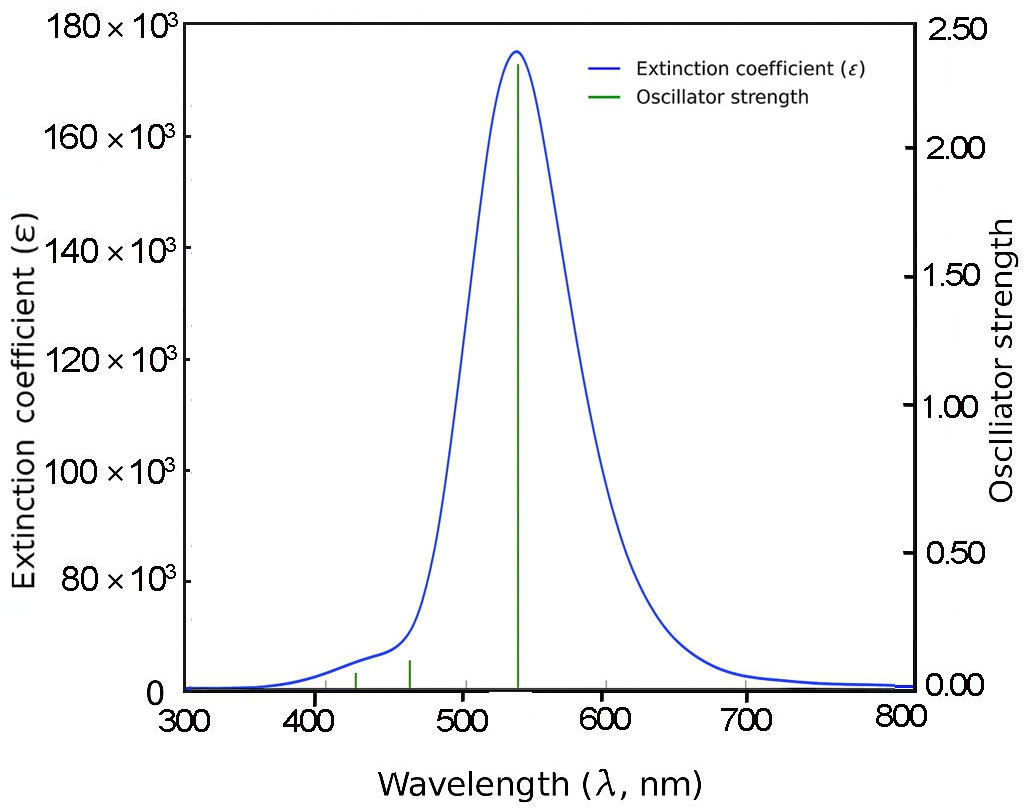
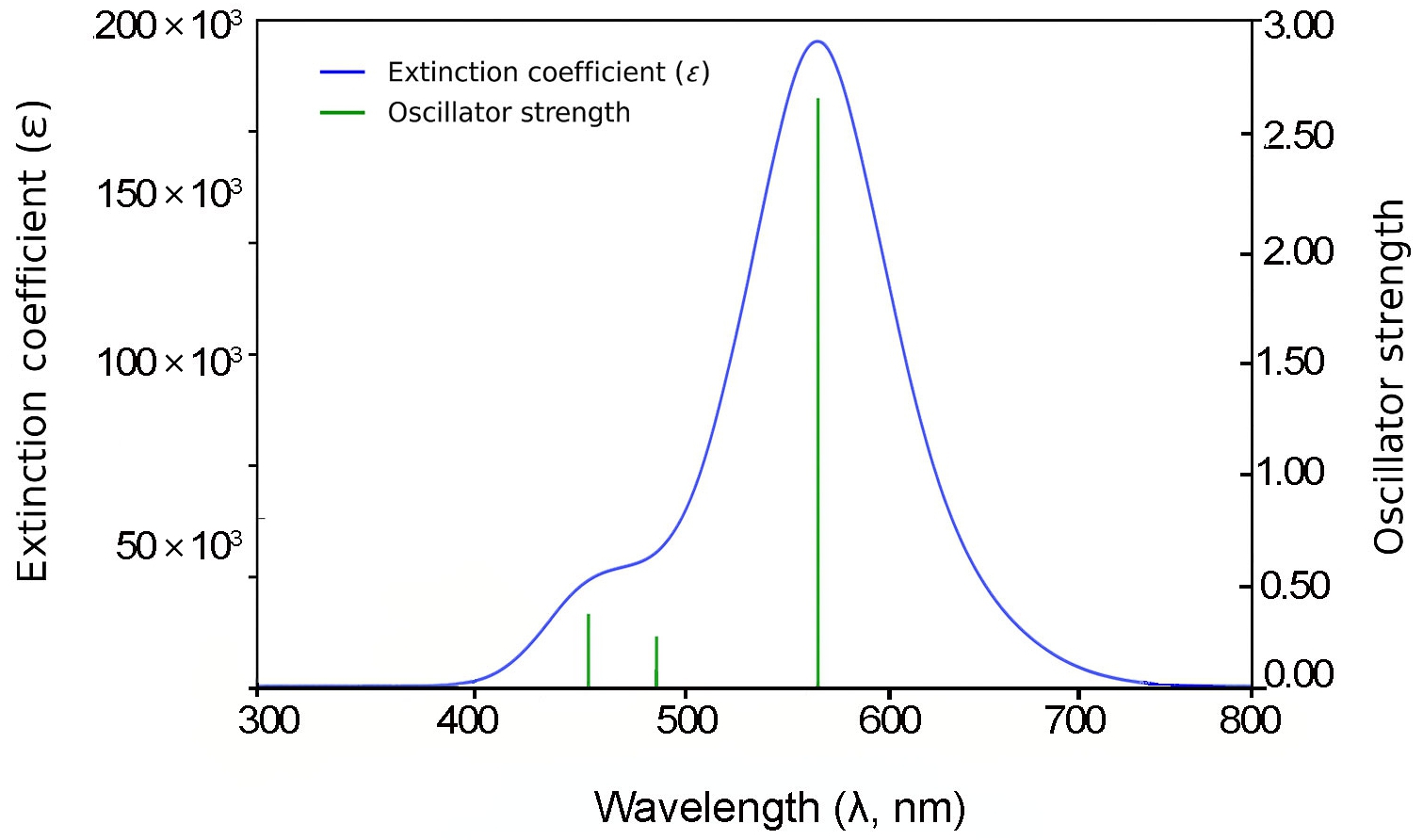
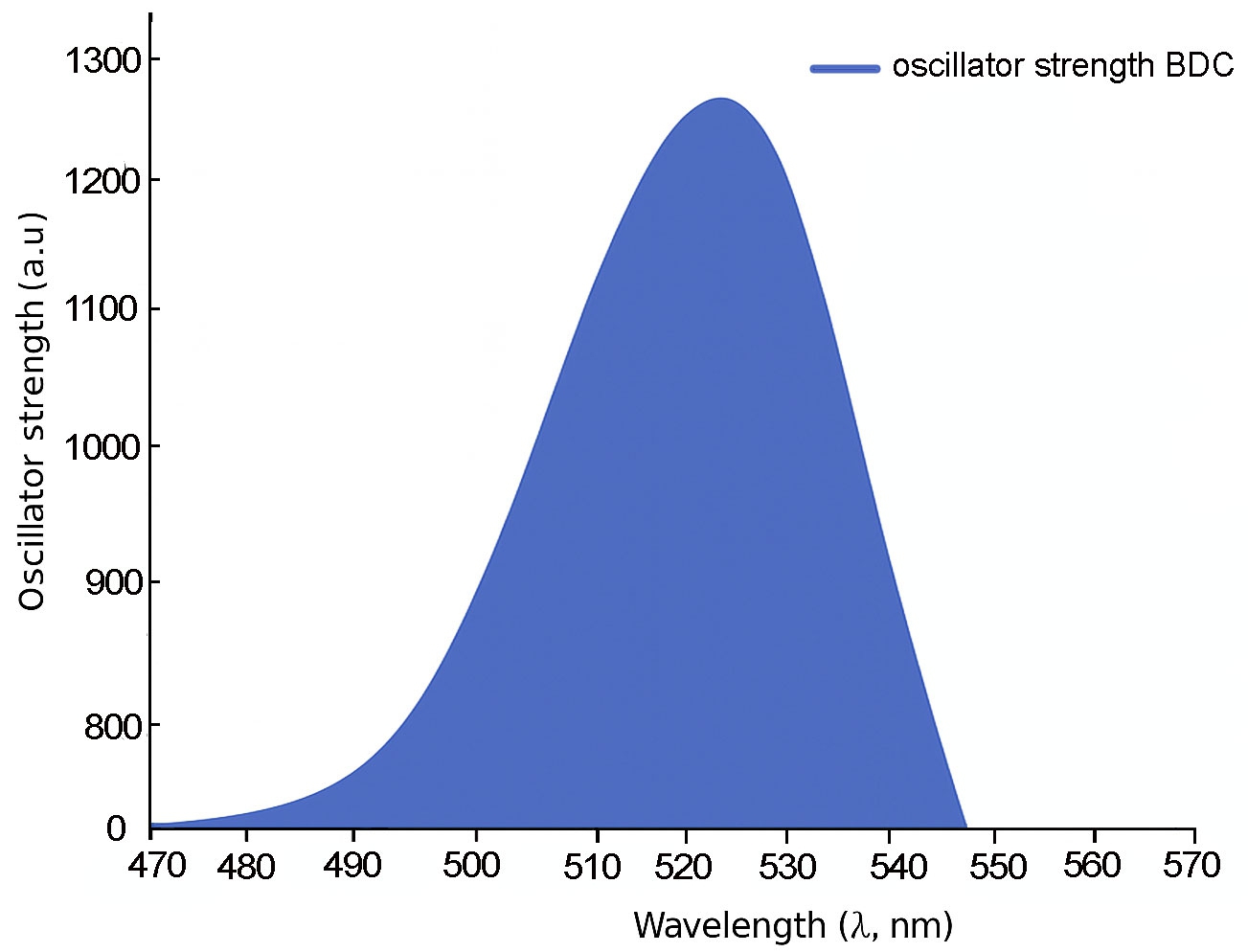
For 3B and 4B (Figs. 5 and 6), further elongation of the π-conjugated system results in additional red shifts of the absorption maxima to 484.65 nm and 531.82 nm, respectively, accompanied by stronger oscillator strengths (1.780 and 2.390). The longest molecule, 5B (Fig. 7), shows the most red-shifted peak at 566.31 nm and the highest oscillator strength of 2.630, confirming a direct correlation between conjugation length and optical activity [60, 61].
Fig. 6. UV-visible absorption spectrum and oscillator strength profile of BDC molecule 4B.
The numerical values of the maximum absorption wavelengths (λmax) and oscillator strengths for all molecules are summarized in Table 3. These results are consistent with the visual trends observed in Figs. 3 to 7 and reinforce the role of molecular extension in enhancing light-matter interactions. The optical transitions for all BDC derivatives are dominated by HOMO → LUMO character, supporting their suitability as active media in nanoscale photonic and optoelectronic systems.
TD-DFT simulated absorption maxima and oscillator strengths for BDC molecules 1B–5B.
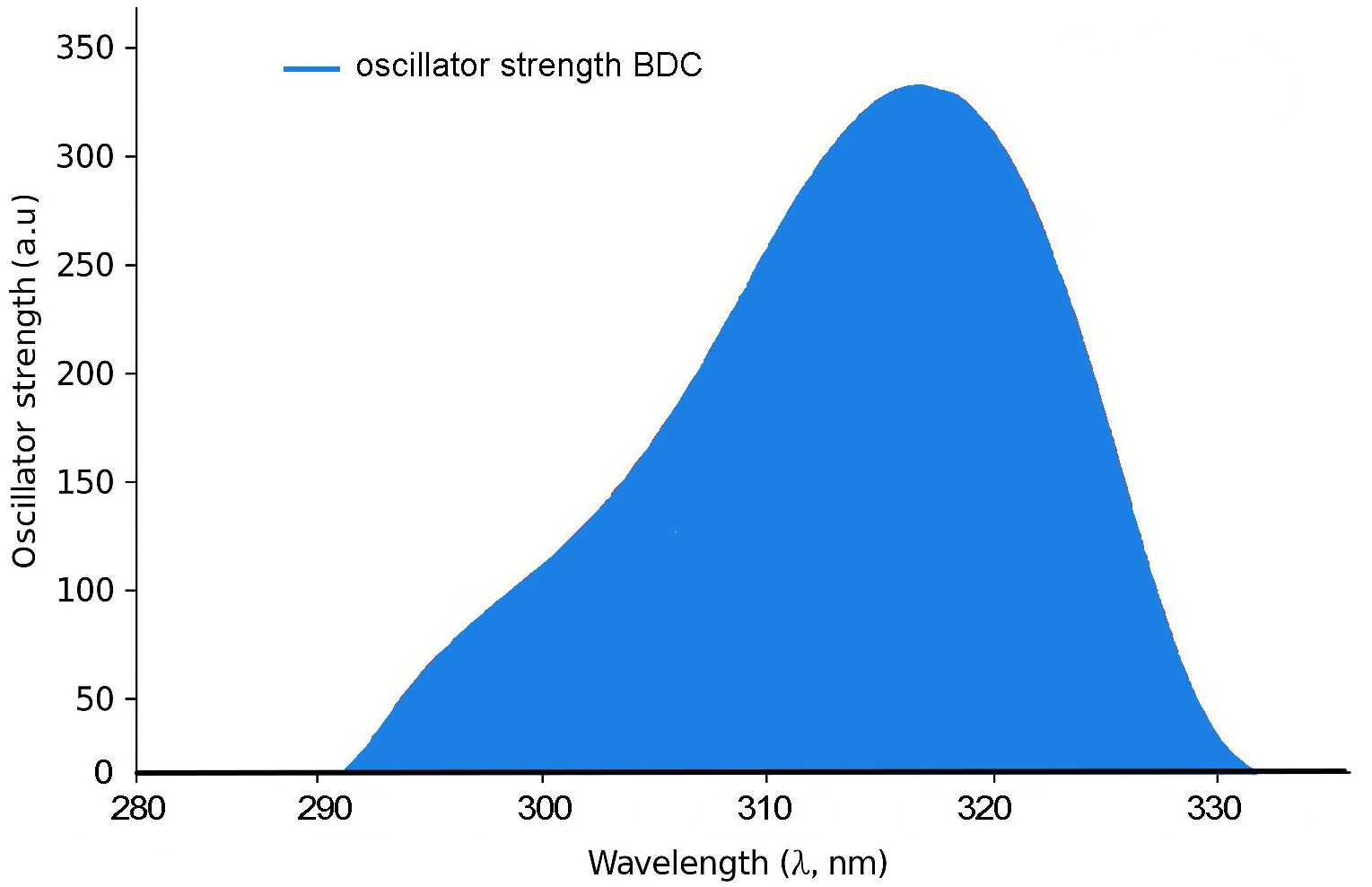
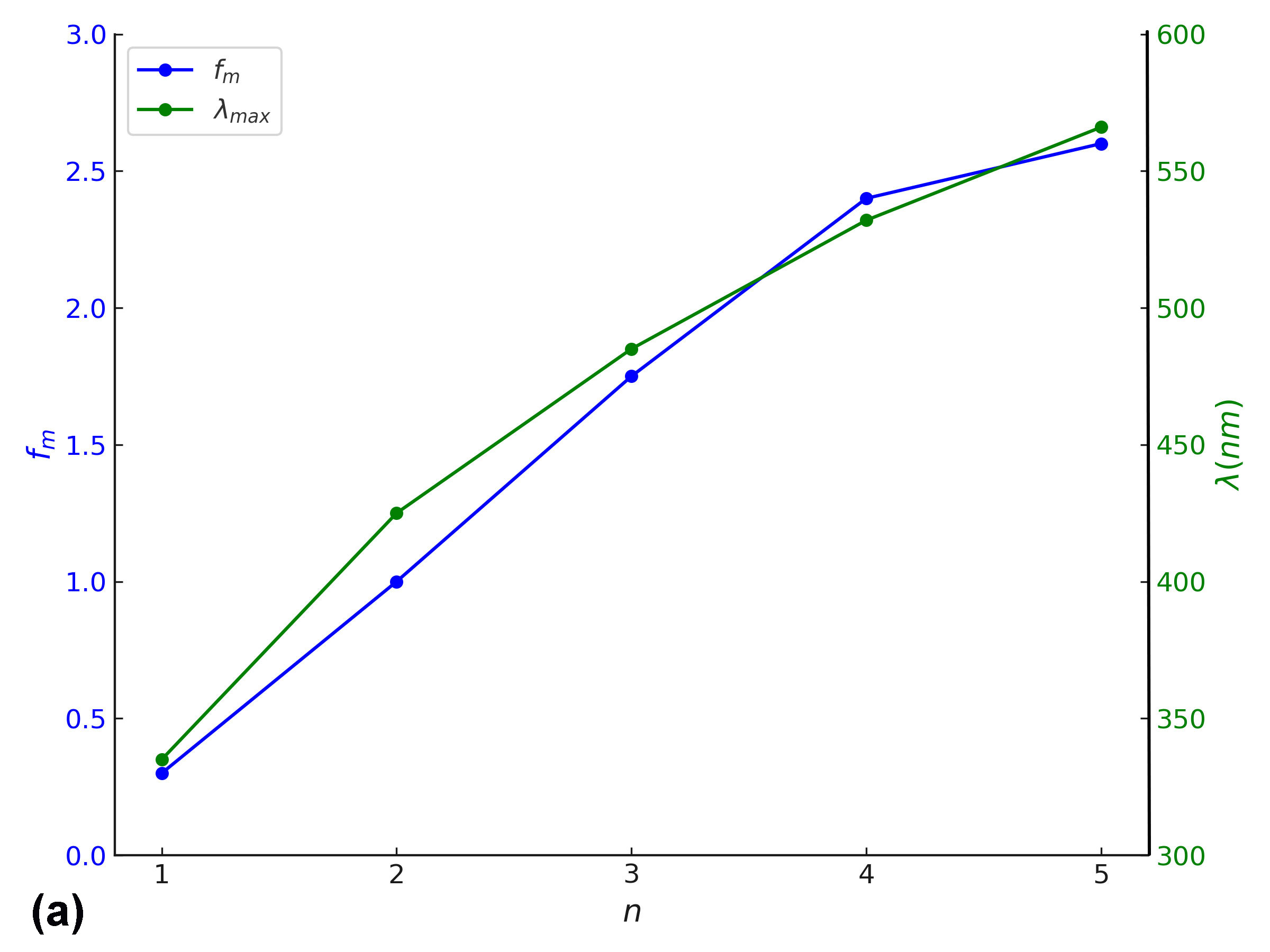
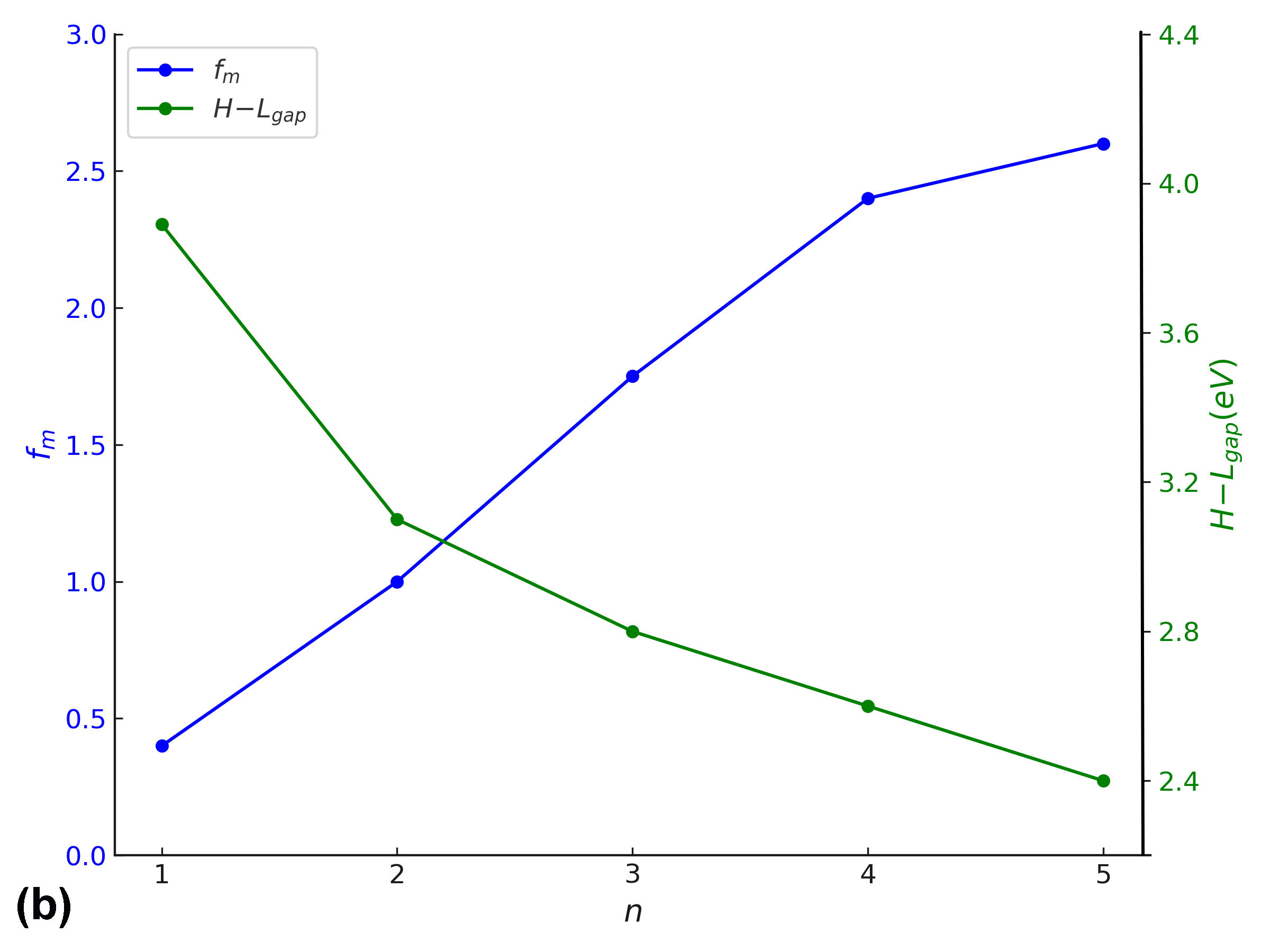
In addition to energy level modulation, a pronounced enhancement in oscillator strength (fₘ) is observed with increasing conjugation length across the BDC molecular series. This trend reflects an increase in the transition dipole moment and radiative transition probability, indicative of stronger light–matter interaction as π-conjugation extends. Such behaviour is crucial for materials intended for optical gain applications, including organic lasers and light-emitting systems [6, 39, 40]. As depicted in Fig. 8(a), both oscillator strength and λₘₐₓ exhibit nearly linear growth with increasing molecular length (n), suggesting that extended conjugation not only intensifies optical transitions but also induces red-shifted absorption [62].
Fig. 8. (a) Variation of oscillator strength (fₘ) and absorption maxima (λₘₐₓ) as a function of conjugation length (n); (b) correlation between
oscillator strength (fₘ) and HOMO–LUMO energy gap (H–Lgap) for BDC molecules with increasing π-conjugation.
Furthermore, Fig. 8(b) illustrates an inverse correlation between oscillator strength and the HOMO–LUMO energy gap, implying that as the electronic bandgap narrows, the transition probability and optical activity increase substantially [63]. These findings confirm that both electronic and optical properties of BDC molecules can be rationally tuned via structural extension, supporting their viability in nanoscale optoelectronic and photonic devices [18].
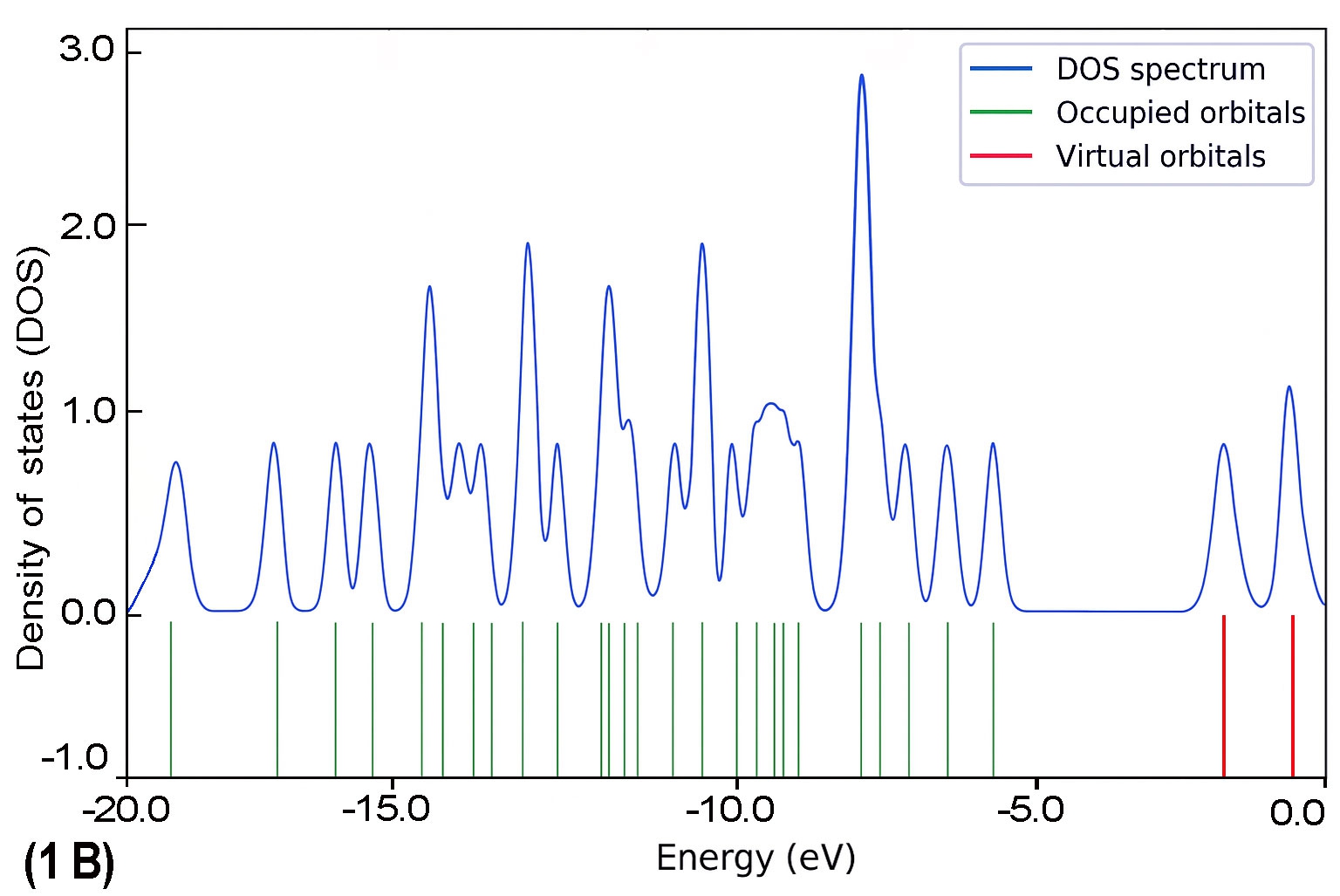
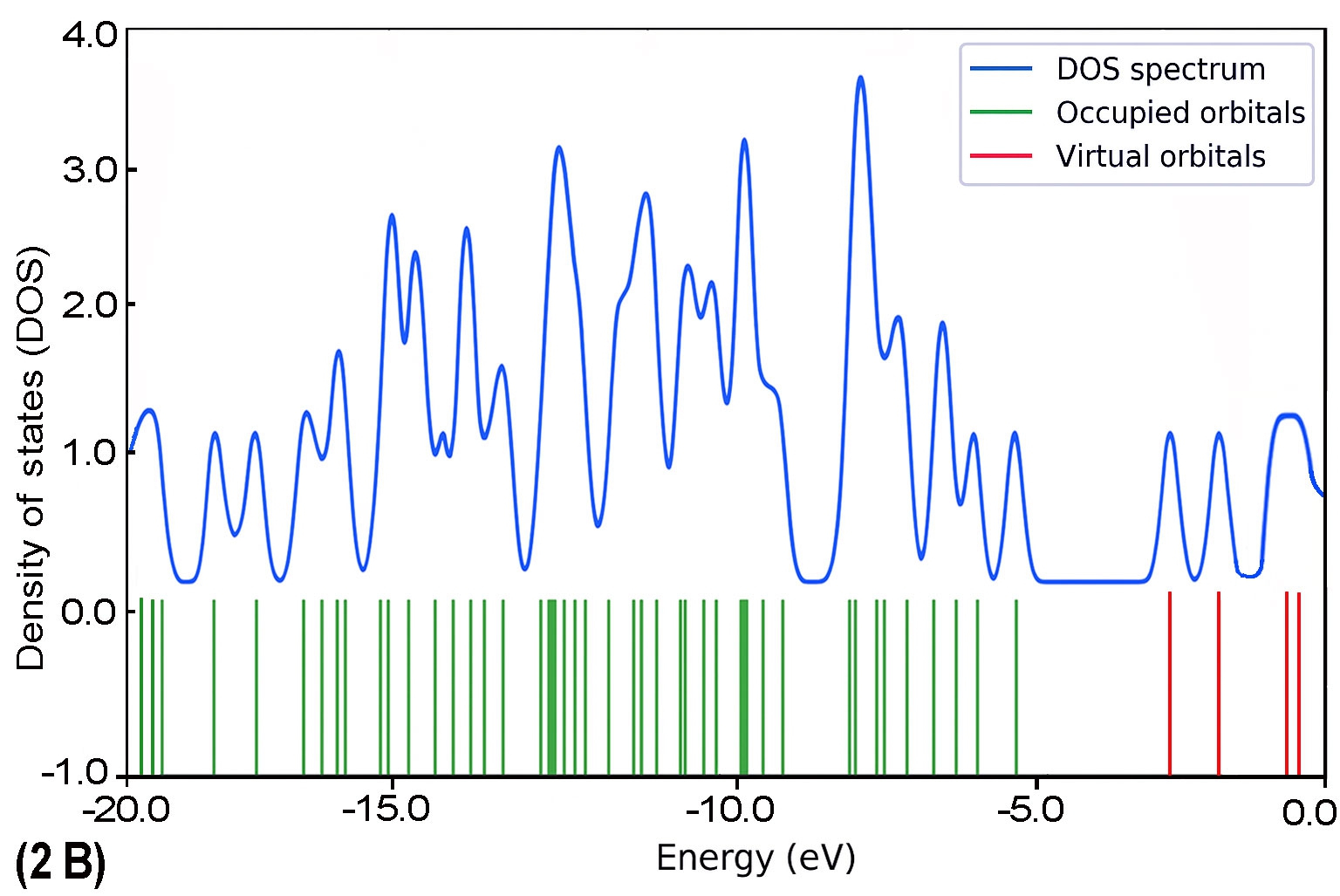
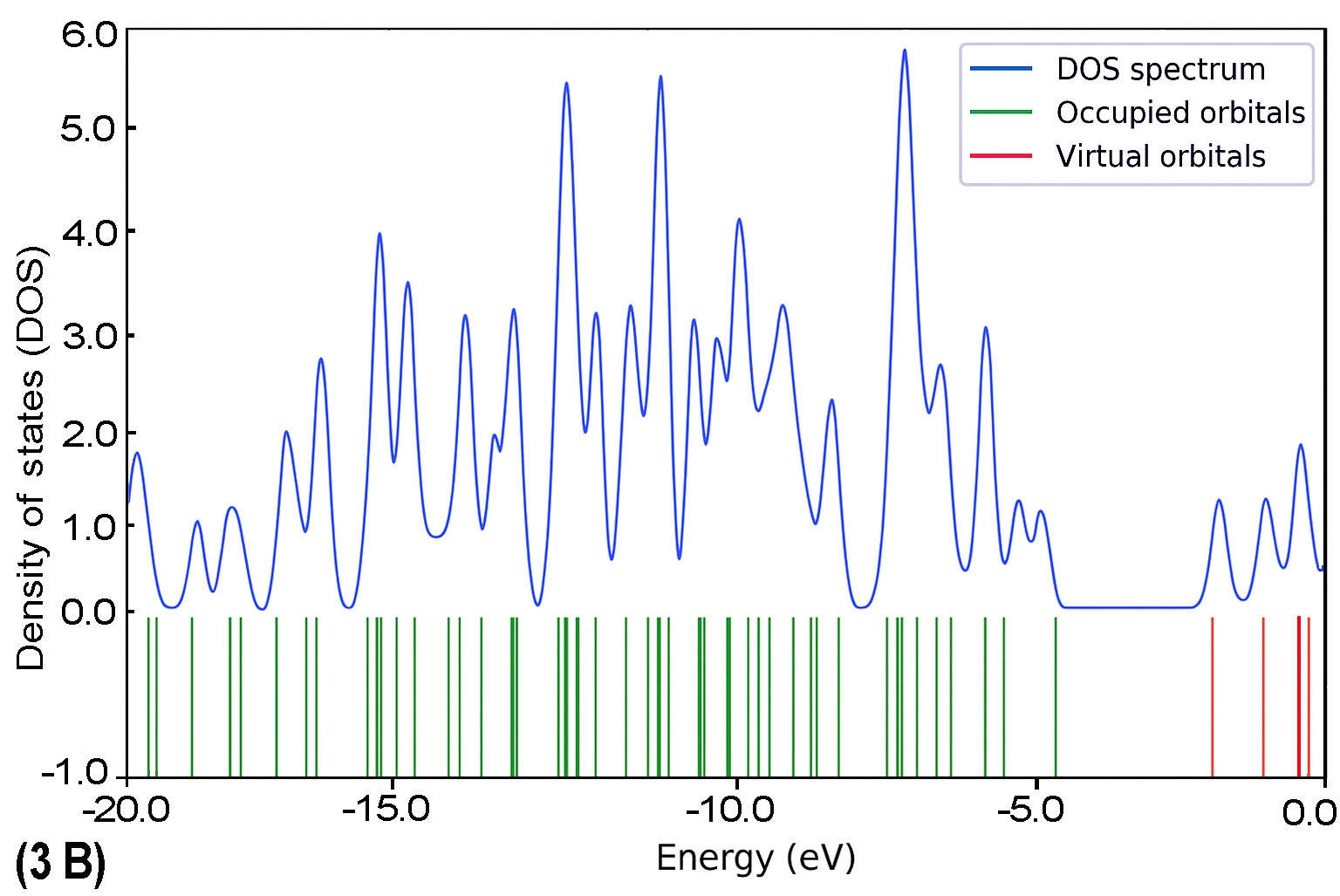
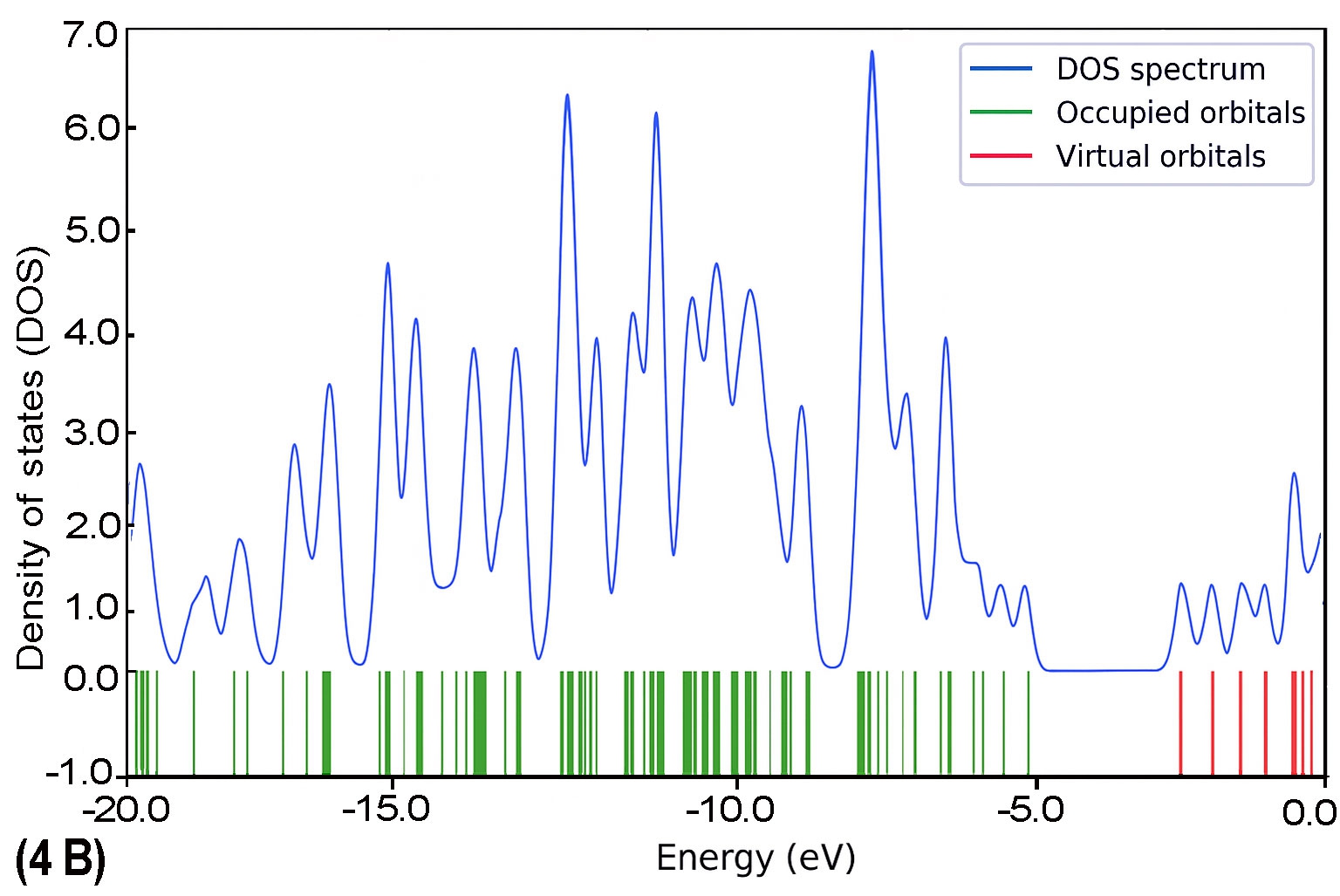
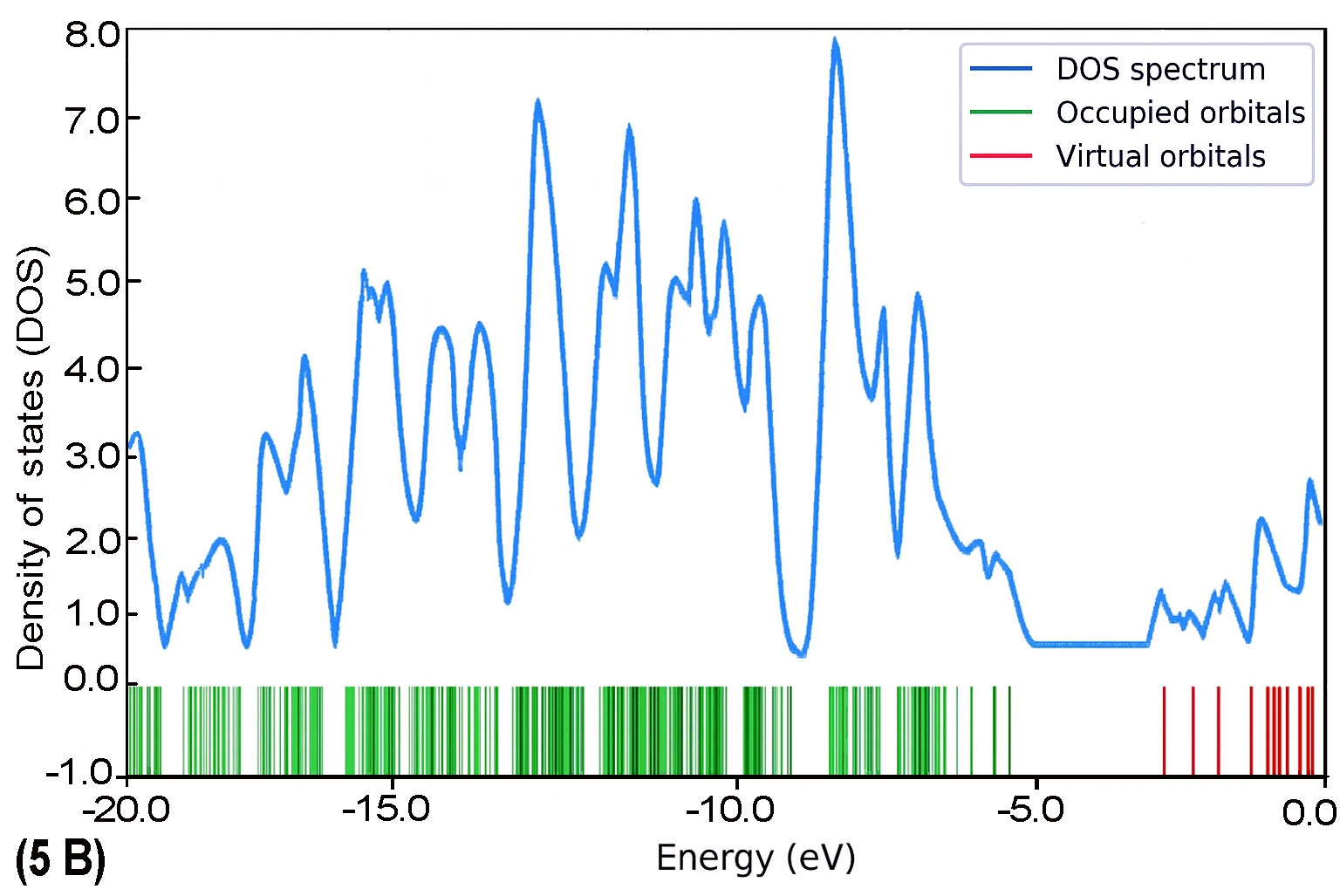
To further explore the electronic characteristics of the BDC molecular series, density of states (DOS) analysis was conducted for each molecule (1B to 5B). This analysis reveals the energy distribution of molecular orbitals and their availability for charge transport. In each DOS spectrum, the blue line represents the total DOS, green lines correspond to occupied orbitals, and red lines indicate virtual (unoccupied) orbitals. A noticeable trend is observed: as the molecular backbone extends, the DOS near the Fermi level (set at 0 eV) becomes denser and more complex. This reflects enhanced electronic delocalization and a narrowing HOMO–LUMO gap. Additionally, the progressive shift in peak distribution from HOMO-dominated to LUMO-dominated regions supports the conclusion of a length-dependent transition in charge transport behaviour.
Density of states (DOS)
To gain deeper insight into the electronic structure of the BDC molecular series, a DOS analysis was performed for each compound. The DOS spectrum reveals the distribution of electronic states across energy levels and provides valuable information on the availability of orbitals for charge transport processes [58,64]. These simulations were conducted within the DFT framework, using the same computational parameters as in the electronic structure and transport studies [46].
The calculated DOS profiles for all BDC derivatives are presented in Fig. 9. Across the series, a clear asymmetry is observed between the occupied and unoccupied electronic states. The occupied states, associated primarily with the
HOMO and deeper orbitals, exhibit dense and sharp peaks,indicating localized electron density. In contrast, the unoccupied states including the LUMO and adjacent orbitals appear more diffuse and energetically broadened, which is consistent with increased delocalization [65].
Fig. 9. DOS spectra of BDC molecules 1B through 5B. Each panel shows the total DOS (blue line), occupied orbitals (green), and virtual
orbitals (red) as a function of energy (eV).
As molecular length increases from 1B to 5B, the DOS near the Fermi level (set at 0 eV) becomes progressively denser. This behaviour reflects enhanced π-conjugation and a consistent reduction in the HOMO–LUMO energy gap [51], both of which are favourable for improving electronic coupling with metal electrodes [27].
As molecular length increases from 1B to 5B, the DOS near the Fermi level (set at 0 eV) becomes progressively denser. This behaviour reflects enhanced π-conjugation and a consistent reduction in the HOMO–LUMO energy gap [51], both of which are favourable for improving electronic coupling with metal electrodes [27].
Moreover, a noticeable shift in the energy alignment of frontier orbitals is observed: while the Fermi level in shorter molecules lies closer to the HOMO, it gradually approaches the LUMO in longer chains. This transition signifies a shift in dominant transport mechanisms from hole-dominated (HOMO-based) to electron-dominated (LUMO-based) conduction as conjugation length increases [66].
As the molecular length of BDC derivatives increases, the density of states near the Fermi level becomes significantly enriched, especially in the LUMO region. This evolution reflects a more delocalized electronic structure and a narrowed HOMO–LUMO energy gap, which enhances electronic coupling with metallic electrodes. In shorter BDC molecules, the DOS near the Fermi level is dominated by HOMO states, favouring hole-mediated transport. However, as conjugation length extends, LUMO states become more energetically aligned with the Fermi level, facilitating a transition to electron-dominated transport.
This shift from HOMO- to LUMO-mediated conduction implies that molecular length can be used as a design parameter to tailor the transport mechanism in nanoelectronic applications. By strategically controlling the conjugation length, it is possible to optimize carrier type, injection efficiency, and conductance, which is vital for the development of high-performance molecular rectifiers, switches, and field-effect transistors (FETs) in organic electronics.
These findings confirm the structural tunability of BDC molecules and underscore their potential as active components in next-generation optoelectronic and nano-electronic devices [67].
Practical considerations and outlook
While this study presents a comprehensive theoretical analysis of the optical and electronic properties of BDC derivatives, certain experimental and practical aspects, such as the influence of molecular defects, material homogeneity, photostability, and operational lifetime, remain beyond the scope of this work. These parameters are critical in determining the real-world viability of organic laser-active materials and should be evaluated through experimental synthesis and device testing [6, 48].
Additionally, the performance of BDC-based systems under high-intensity optical excitation, such as resistance to photodegradation or nonlinear effects, requires time-resolved and photophysical characterization, which could further validate the lasing potential predicted computationally [49, 50].
Given their strong absorption, large oscillator strengths, and tunable electronic structure, the BDC derivatives studied here may be suited for use in organic distributed feedback (DFB) lasers or vertical cavity surface-emitting lasers (VCSELs) operating in the visible to near-infrared range. These applications warrant future work involving experimental fabrication and laser threshold analysis [51, 52].
Impact of molecular length on transport mechanisms
The molecular length in BDC derivatives plays a pivotal role in determining the dominant transport mechanism as it directly influences the spatial distribution and energy alignment of the frontier molecular orbitals (HOMO and LUMO) relative to the Fermi level of the electrodes. In shorter BDC molecules, the HOMO levels lie closer to the Fermi energy, and the HOMO orbitals are more localized near the anchoring heteroatoms, facilitating hole-mediated transport [68]. However, as the molecular backbone is extended, π-conjugation increases, leading to greater delocalization of the LUMO orbitals across the conjugated carbon framework and a simultaneous reduction in the HOMO–LUMO energy gap [69, 70].
This evolution results in the LUMO energy level gradually shifting closer to the electrode Fermi level, promoting a transition from HOMO-dominated (hole) conduction to LUMO-dominated (electron) conduction in longer molecules [71]. Such a length-dependent transition in transport behaviour provides a powerful structural handle for tuning charge injection and transmission characteristics in molecular junctions.
From a device design perspective, this property enables the development of length-tunable nanoelectronic compo-nents, where the conduction type (p-type vs. n-type) and current magnitude can be modulated by controlling the conjugation length of the molecular bridge. This tunability is particularly advantageous in the context of molecular rectifiers, switches, and organic field-effect transistors (OFETs), where the type and mobility of charge carriers are critical for device performance [72, 73].
Conclusions
This study presents a comprehensive theoretical analysis of the structural, electronic, optical, and charge transport properties of BDC molecules, evaluated as potential single-molecule active media for nanoscale optoelectronic and photonic applications. Through the integration of DFT, TD-DFT, and NEGF formalism, the authors systematically examined the influence of molecular length and π‑conjugation on key electronic and photophysical charac-teristics.
The authors’ findings reveal that an increasing molecular length results in a consistent narrowing of the HOMO–LUMO energy gap, enhanced orbital delo-calization, and improved alignment of frontier orbitals with the electrode Fermi levels. These effects collectively lead to more efficient charge transport and stronger optical responses, both of which are essential attributes for active laser components and molecular-scale optoelectronic devices.
TD-DFT simulations of the absorption spectra revealed a clear red shift in excitation energies and a substantial increase in oscillator strengths across the BDC series, confirming the strengthening of light–matter interaction with extended conjugation. These optical properties, particularly the increasing oscillator strength and bathochromic shift in absorption, demonstrate strong light–matter interaction and are characteristic of systems with potential for radiative transitions. While laser performance metrics, such as optical gain or emission cross-section, were not directly calculated, the observed spectral behaviour supports the feasibility of BDC molecules as promising candidates for the future development of organic laser-active materials.
Additionally, a DOS analysis revealed a notable transition in transport behaviour: from HOMO-dominated (hole-mediated) conduction in shorter molecules to LUMO-dominated (electron-mediated) conduction in longer structures. This transition highlights the tunability of charge transport mechanisms in relation to molecular architecture.
In conclusion, this work highlights the effectiveness of molecular-scale structural engineering in tailoring optoelectronic properties. BDC derivatives, with their planar rigid backbones and length-dependent conjugation, emerge as up-and-coming candidates for the design of next-generation organic lasers and nanoelectronic components. Future efforts should focus on experimental validation and the practical integration of these molecular systems into scalable device platforms.
Ethics approval and consent to participate
All procedures were performed in accordance with the ethical standards of the institutional and national research committees, as well as the Declaration of Helsinki. This study was approved by the Research Ethics Committee of the Faculty of Health and Medical Techniques at Al‑Mustaqbal University on 10/03/2025 (reference number 10032025)
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
The authors sincerely thank the Deanship of the College of Health and Medical Techniques at Al-Mustaqbal University for their valuable assistance in data collection.
References
Avouris, P., Chen, Z. & Perebeinos, V. Carbon-based electronics. Nat. Nanotechnol. 2, 605–615 (2007). https://doi.org/10.1038/nnano.2007.300
Javey, A., Guo, J., Wang, Q., Lundstrom, M. & Da i, H. Ba llistic carbon nanotube field-effect transistors. Nature 424, 654–657 (2003). https://doi.org/10.1038/nature01797
Geim, A. K. & Novoselov, K. S. The Rise of Graphene. in Nanoscience and Technology. A Collection of Reviews from Nature Journals (ed. Rodgers, P.) 11–19 (Nature Publishing Group, 2009)https://doi.org/10.1142/9789814287005_0002
Anthony, J. E. Functionalized acenes and heteroacenes for organic electronics. Chem. Rev. 106, 5028–5048 (2006) https://doi.org/10.1021/cr050966z
Wu, X. et al. An oriented design of a π-conjugated polymer framework for high-performance solid-state lithium batteries. Energy Environ. Sci. 18, 1835–1846 (2025).https://doi.org/10.1039/d4ee03104k
Cachaneski-Lopes, J. P. & Batagin-Neto, A. Effects of mechanical deformation on the opto-electronic responses, reactivity, and performance of conjugated polymers: A DFT study. Polymers (Basel) 14, 1354 (2022). https://doi.org/10.3390/polym14071354
Matsuda, M. et al. Impact of the heteroatoms on mobility- stretchability properties of n-type semiconducting polymers with conjugation break spacers. Macromolecules 56, 2348–2361 (2023).https://doi.org/10.1021/acs.macromol.3c00109
Peng, X. et al. Construction frontier molecular orbital prediction model with transfer learning for organic materials. npj Comput.Mater. 10, 213 (2024). https://doi.org/10.1038/s41524-024-01403-6
Kha lid, M. et al. Exploration of second and third order nonlinear optical properties for theoretical framework of organic D–π–D–π– A type compounds. Opt. Quantum Electron. 53, 561 (2021). https://doi.org/10.1007/s11082-021-03212-3
Quochi, F. et al. Extending the lasing wavelength coverage of organic semiconductor nanofibers by periodic organic-organic heteroepitaxy. Adv. Opt. Mater. 1, 117–122 (2013). https://doi.org/10.1002/adom.201200005
Loudet, A. & Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 107, 4891– 4932 (2007). https://doi.org/10.1021/cr078381n
Tang, M. L. & Bao, Z. Halogenated materials as organic semiconductors. Chem. Mater. 23, 446–455 (2011).https://doi.org/10.1021/cm102182x
Li, Y. & Zou, Y. Conjugated polymer photovoltaic materials with broad absorption band and high charge carrier mobility. Adv. Mater. 20, 2952–2958 (2008). https://do.org/10.1002/adma.200800606
Gierschner, J. & Pa rk, S. Y. Luminescent distyrylbenzenes: Tailoring molecular structure and crystalline morphology. J. Mater.>Chem. C 1, 5818–5832 (2013). https://doi.org/10.1039/c3tc31062k
Scherf, U., Riechel, S., Lemmer, U. & Mahrt, R. F. Conjugated polymers: Lasing and stimulated emission. Curr. Opin. Solid State Mater. Sci. 5, 143–154 (2001). https://doi.org/10.1016/S1359-0286(01)00010-9
Friederich, P. et al. Molecular origin of the charge carrier mobility in small molecule organic semiconductors. Adv. Funct. Mater. 26, 5757–5763 (2016). https://doi.org/10.1002/adfm.201601807
Maadhu, T. & Gandhiraj, V. Design, crystal growth and characterizations of novel bis morpholinium zinc bromide single crystals for optoelectronic applications. Opt. Mater. 138 , 113694 (2023). >https://doi.org/10.1016/j.optmat.2023.113694
Maadhu, T. et al. Novel metal – organic framework and crystal engineering of bismorpholinium mercury(II) tribromo chloride (BMMC) for optoelectronic applications. Cryst. Growth Des. 24 , 1632–1647 (2024). https://doi.org/10.1021/acs.cgd.3c01284
Maadhu, T., Gopalakrishnan, A., Gandhiraj, V. & Senthil Pandian,M. Physicochemical properties of 2-amino-5-methylpyridinium 3- carboxy-4-hydroxybenzenesulfonate single crystal: An efficient material for optoelectronic applications. J. Mol. Struct. 1319, 139536 (2025). https://doi.org/10.1016/j.molstruc.2024.139536
Maadhu, T. et al. Synthesis, experimental and theoretical studies of morpholinium bromide single crystal for NLO application. J. Mol. Struct. 1294, 136522 (2023). https://doi.org/10.1016/j.molstruc.2023.136522
Kharissova, O. V., Kharisov, B. I. & González, L. T. Recent trends on density functional theory-assisted calculations of structures and properties of metal-organic frameworks and metal-organic frameworks-derived nanocarbons. J. Mater. Res. 35, 1424–1438 (2020). https://doi.org/10.1557/jmr.2020.109
Shi, W., Wang, D. & Shuai, Z. High‐performance organic thermoelectric materials: Theoretical insights and computational design. Adv. Electron. Mater. 5, 1800882 (2019).https://doi.org/10.1002/aelm.201800882
Lambert, C. J. Quantum Transport in Nanostructures and Molecules: An Introduction to Molecular Electronics. (IoP Publishing, 2021).
García Arriba s, A. et al. Siesta: Recent developments and applications. J. Chem. Phys. 152, 204208 (2020). https://doi.org/10.1063/5.0005077
Yu, Y. et al. A p-π* conjugated triarylborane as an alcohol- processable n-type semiconductor for organic optoelectronic devices. J. Mater. Chem. C 7, 7427–7432 (2019). https://doi.org/10.1039/C9TC01562K
Becke, A. D. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993). https://doi.org/10.1063/1.464913
Cuevas, J. C. & Scheer, E. Molecular Electronics: An Introduction To Theory and Experiment. (World Scientific, 2010).
Adamo, C. & Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999). https://doi.org/10.1063/1.478522
Teunissen, J. L., De Proft, F. & De Vleeschouwer, F. Tuning the HOMO–LUMO energy gap of small diamondoids using inverse molecular design. J. Chem. Theory Comput. 13, 1351–1365 (2017). https://doi.org/10.1021/acs.jctc.6b01074
Wen, L., Luo, D., Cheng, L., Zhao, K. & Ma, H. Electronic structure properties of two-dimensional π-conjugated polymers. Macromolecules 49, 1305–1312 (2016). https://doi.org/10.1021/acs.macromol.5b02572
Ya ng, Y. et al. Charge transfer and photophysical properties of DSSCs based on different π-conjugated bridges: DFT and TD-DFT study. J. Mol. Graph. Model. 137, 108986 (2025). https://doi.org/10.1016/j.jmgm.2025.108986
Venkataraman, L., Klare, J. E., Nuckolls, C., Hybertsen, M. S. & Steigerwa ld, M. L. Dependence of single-molecule junction conductance on molecular conformation. Nature 442, 904–907 (2006).https://doi.org/10.1038/nature05037
Facchetti, A. π-conjugated polymers for organic electronics and photovoltaic cell applications. Chem. Mater. 23, 733–758 (2011). https://doi.org/10.1021/cm102419z
Oberhofer, H., Reuter, K. & Blumberger, J. Charge transport in molecula r materia ls: An assessment of computational methods. Chem. Rev. 117, 10319–10357 (2017). https://doi.org/10.1021/acs.chemrev.7b00086
Moth-Poulsen, K. & Bjørnholm, T. Molecular electronics with single molecules in solid-sta te devices. Nat. Nanotechnol. 4, 551– 556 (2009).https://doi.org/10.1038/nnano.2009.176
Bahari, Y., Mortazavi, B., Rajabpour, A., Zhuang, X. & Rabczuk, T. Application of two-dimensional materials as anodes for rechargeable metal-ion batteries: A comprehensive perspective from density functional theory simulations. Energy Storage Mater. 35, 203–282 (2021). https://doi.org/10.1016/j.ensm.2020.11.004
Rubert-Albiol, R. et al. Charge transport in structurally related organic semiconductors: experimental determination and computational modeling in crystalline and amorphous scenarios. J. Phys. Chem. C 129, 5960–5972 (2025). https://doi.org/10.1021/acs.jpcc.4c08068
Aradhya, S. V. & Venkataraman, L. Single-molecule junctions beyond electronic transport. Nat. Nanotechnol. 8, 399–410 (2013).https://doi.org/10.1038/nnano.2013.91
Quek, S. Y. et al. Mechanically controlled binary conductance switching of a single-molecule junction. Nat. Nanotechnol. 4, 230– 234 (2009). https://doi.org/10.1038/nnano.2009.10
Sun, L. et al. Single-molecule electronics: From chemical design to functional devices. Chem. Soc. Rev. 43, 7378–7411 (2014). https://doi.org/10.1039/C4CS00143E
Anthony, J. E. Functionalized acenes and heteroacenes for organic electronics. Chem. Rev. 106, 5028–5048 (2006). https://doi.org/10.1021/cr050966z
Bhat,V., Callaway, C. P. & Risko, C. Computational approaches for organic semiconductors: From chemical and physical understanding to predicting new materials. Chem. Rev. 123, 7498– 7547 (2023). https://doi.org/10.1021/acs.chemrev.2c00704
Xu, B. & Tao, N. J. Measurement of single-molecule resistance by repeated formation of molecular junctions. Science 301, 1221– 1223 (2003). https://doi.org/10.1126/science.108748
Liu, H. et al. Length‐dependent conductance of molecular wires and contact resistance in metal–molecule–meta l junctions. ChemPhysChem 9, 1416–1424 (2008). https://doi.org/10.1002/cphc.200800032
Yasini, P. et al. Potential-induced high-conductance transport pathways through single-molecule junctions. J. Am. Chem. Soc.141 , 10109–10116 (2019). https://doi.org/10.1021/jacs.9b05448
Khoo, K. H., Chen, Y., Li, S. & Quek, S. Y. Length dependence of electron transport through molecular wires – a first principles perspective. Phys. Chem. Chem. Phys. 17, 77–96 (2015). https://doi.org/10.1039/C4CP05006A
Lambert, C. J. Basic concepts of quantum interference and electron transport in single-molecule electronics. Chem. Soc. Rev. 44, 875– 888 (2015). https://doi.org/10.1039/C4CS00203B
Dreuw, A. & Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 105, 4009–4037 (2005).https://doi.org/10.1021/cr0505627
Spano, F. C. The spectral signatures of Frenkel polarons in H-and J-aggrega tes. Acc. Chem. Res. 43, 429–439 (2010). https://doi.org/10.1021/ar900233v
Zeng, X. Y. et al. Extended conjugation strategy enabling red- shifted and efficient emission of orange-red thermally activated delayed fluorescence materials. ACS Appl. Mater. Interfaces 16, 16563–16572 (2024). https://doi.org/10.1021/acsami.3c18880
Yang, et al. Charge transfer and photophysical properties of DSSCs based on different π-conjugated bridges: DFT and TD-DFT study. J. Mol. Graph. Model. 137, 108986 (2025). https://doi.org/10.1016/j.jmgm.2025.108986
Datta, S. Electronic Transport in Mesoscopic Systems. (Cambridge University Press, 1997).
Quek, S. Y. et al. Amine – gold linked single-molecule circuits: Experiment and theory. Nano Lett. 7, 3477–3482 (2007). https://doi.org/10.1021/nl072058i
Xia ng, D., Wang, X., Jia , C., Lee, T. & Guo, X. Molecula r-scale electronics: From concept to function. Chem. Rev. 116, 4318–4440 (2016). https://doi.org/10.1021/acs.chemrev.5b00680
Chen, H. et al. Single-molecule charge transport through positively charged electrostatic anchors. J. Am. Chem. Soc. 143, 2886–2895 (2021). https://doi.org/10.1021/jacs.0c12664
Khoo, K. H., Chen, Y., Li, S. & Quek, S. Y. Length dependence of electron transport through molecular wires – a first principles perspective. Phys. Chem. Chem. Phys. 17, 77–96 (2015). https://doi.org/10.1039/c4cp05006a
Feng, S.-W., Shih, M.-C., Huang, C. J. & Chung, C.-T. Impacts of dopant concentration on the carrier transport and recombination dynamics in organic light emitting diodes. Thin Solid Films 517, 2719–2723 (2009). https://doi.org/10.1016/j.tsf.2008.10.049
Smith, C. E. et al. Length-dependent nanotransport and charge hopping bottlenecks in long thiophene-containing π-conjugated molecula r wires. J. Am. Chem. Soc. 137, 15732–15741 (2015). https://doi.org/10.1021/jacs.5b07400
Sirringhaus, H., Bird, M., Richards, T. & Zhao, N. Charge transport physics of conjugated polymer field-effect transistors. Adv. Mater. 22, 3893–3898 (2010). https://doi.org/10.1002/adma.200902857
Ja sim, S. A. et al. Molecular junctions: Introduction and physical foundations, nanoelectrical conductivity and electronic structure and charge transfer in organic molecular junctions. Braz. J. Phys. 52, 31 (2022). https://doi.org/10.1007/s13538-021-01033-z